医学遗传学
-
1.1目录
-
1.2序言
-
1.3前言
-
1.4第一章 绪论
-
1.4.1第一节 医学遗传学的任务和范畴
-
1.4.2第二节 医学遗传学发展简史
-
1.4.3第三节 遗传病概述
-
1.4.4第四节 遗传病的研究策略
-
1.5第二章 基因突变
-
1.5.1第一节 诱发基因突变的因素
-
1.5.2第二节 基因突变的一般特性
-
1.5.3第三节 基因突变的分子机制
-
1.5.4第四节 DNA损伤的修复
-
1.6第三章 突变基因的分子细胞生物学效应
-
1.6.1第一节 基因突变导致蛋白质功能改变
-
1.6.2第二节 基因突变引起性状改变的细胞生物学机制
-
1.7第四章 单基因疾病的遗传
-
1.7.1第一节 系谱与系谱分析法
-
1.7.2第二节 常染色体显性遗传病的遗传
-
1.7.3第三节 常染色体隐性遗传病的遗传
-
1.7.4第四节 X连锁显性遗传病的遗传
-
1.7.5第五节 X连锁隐性遗传病的遗传
-
1.7.6第六节 Y连锁遗传病的遗传
-
1.7.7第七节 影响单基因遗传病分析的几个因素
-
1.8第五章 单基因遗传病
-
1.8.1第一节 分子病
-
1.8.2第二节 先天性代谢病
-
1.9第六章 疾病的多基因遗传
-
1.9.1第一节 数量性状的多基因遗传
-
1.9.2第二节 多基因遗传病的遗传
-
1.10第七章 多基因遗传病
-
1.10.1第一节 精神分裂症
-
1.10.2第二节 糖尿病
-
1.10.3第三节 哮喘
-
1.10.4第四节 原发性高血压
-
1.11第八章 线粒体遗传
-
1.11.1第一节 人类线粒体基因组
-
1.11.2第二节 线粒体基因的突变
-
1.11.3第三节 线粒体遗传的特点
-
1.12第九章 线粒体疾病
-
1.12.1第一节 疾病过程中的线粒体变化
-
1.12.2第二节 mt DNA突变引起的疾病
-
1.13第十章 人类染色体
-
1.13.1第一节 人类染色体的基本特征
-
1.13.2第二节 染色体分组、核型与显带技术
-
1.14第十一章 染色体畸变
-
1.14.1第一节 染色体畸变发生的原因
-
1.14.2第二节 染色体数目异常及其产生机制
-
1.14.3第三节 染色体结构畸变及其产生机制
-
1.15第十二章 畸变染色体引起的疾病
-
1.15.1第一节 染色体病发病概况
-
1.15.2第二节 常染色体病
-
1.15.3第三节 Down综合征
-
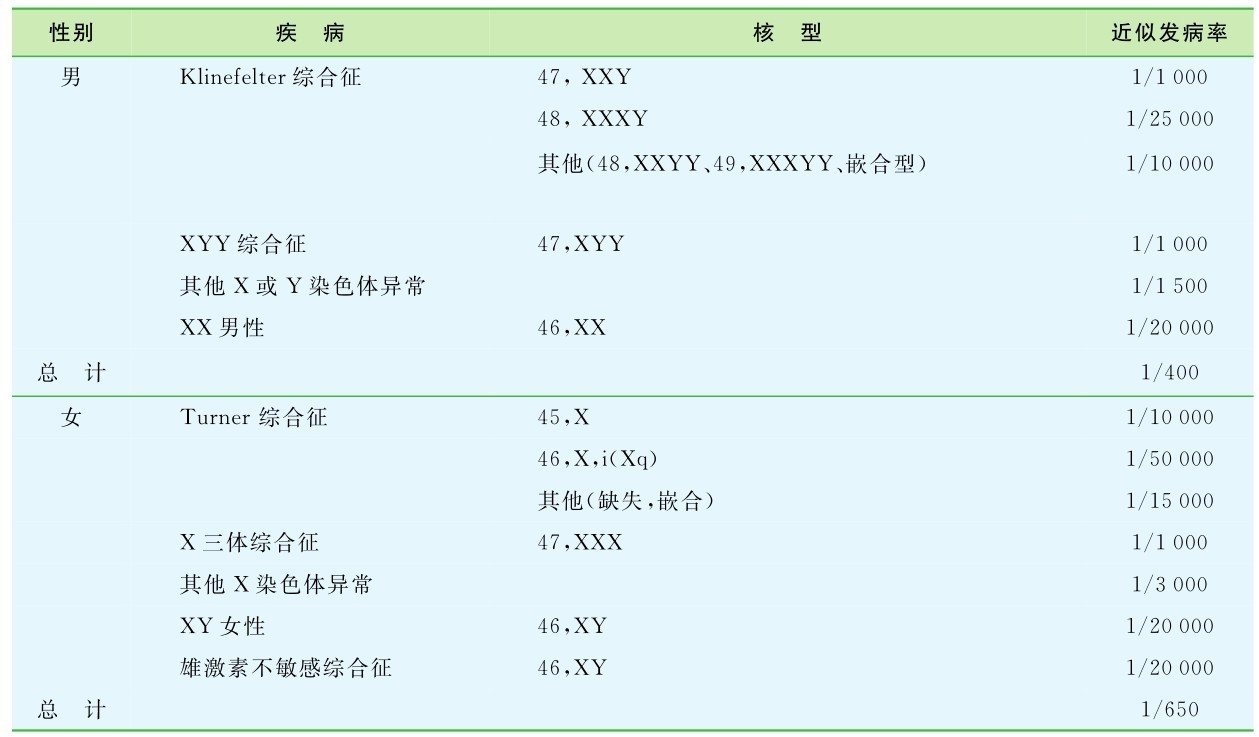
1.15.4第四节 性染色体病
-
1.15.5第五节 染色体异常携带者
-
1.16第十三章 肿瘤与遗传
-
1.16.1第一节 肿瘤发生的遗传现象
-
1.16.2第二节 肿瘤基因
-
1.16.3第三节 肿瘤的多步骤发生和癌基因组解剖计划
-
1.17第十四章 出生缺陷
-
1.17.1第一节 出生缺陷的发生率
-
1.17.2第二节 出生缺陷的临床特征
-
1.17.3第三节 常见的出生缺陷
-
1.17.4第四节 出生缺陷的病理生理学
-
1.18第十五章 表观遗传与疾病
-
1.18.1第一节 表观遗传概述
-
1.18.2第二节 肿瘤中的表观遗传变异
-
1.18.3第三节 神经发育失调疾病与表观遗传
-
1.18.4第四节 糖尿病和肥胖中的表观遗传
-
1.18.5第五节 表观遗传与衰老及干细胞治疗
-
1.19第十六章 遗传病的诊断与治疗
-
1.19.1第一节 遗传病的诊断
-
1.19.2第二节 遗传病的治疗
-
1.20中英文名词对照索引
-
1.21参考文献