
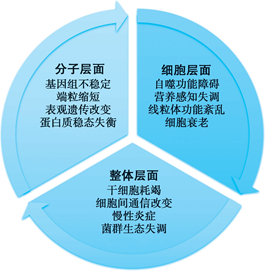
目前已经明确的 12 个衰老标志(hallmarks of aging),根据其起源及作用方式可分为三类,即分子层面、细胞层面、整体层面(图 5- 2)。三个层面的 12个衰老标志并非完全独立,而是相互作用、相互影响,涉及分子、细胞和整体的复杂过程,在衰老发生发展过程中起到重要的促进作用。各衰老标志无论是在实验动物,还是在人类医学研究中,都是对衰老生物体的形态和功能衰退的客观定量,对于测量生物老化至关重要。
一、分子层面衰老标志
(一)基因组不稳定(geneticinstability)
DNA是遗传信息的载体,在细胞复制、人体衰老的过程中,其完整性和稳定性不断受到外源性因素(物理、化学、生物等)和内源性因素(DNA复制错误、自发水解反应、氧化应激等)的影响,引起点突变、缺失、异位、端粒缩短、单链和双链断裂、染色体重排、核结构缺陷等破坏。尽管生物体已经进化出一系列修复核DNA和线粒体 DNA损伤的机制,从而确保染色体维持正常的结构和功能,但这些DNA修复系统会随着年龄的增长而降低甚至失去效率,造成损伤基因组的积累。
老年人细胞及体外培养的衰老细胞的核 DNA上积累了大量的突变。而其他形式的损伤,如染色体非整倍体和拷贝数变异,都会影响基因本身的转录和转录调控途径,导致细胞功能障碍,最终损伤组织、破坏有机体的稳态。DNA损伤后细胞发生分子级联反应,称为DNA损伤反应(DNAdamage reaction,DDR)。DDR激活细胞周期检查点,启动 DNA修复,根据修复结果决定受损细胞的命运。随着年龄的增长,关键的 DNA修复因子和相关蛋白的表达降低,修复因子的募集迟缓,修复保真度下降,导致 DNA修复能力逐渐衰退,核 DNA、线粒体 DNA(mtDNA)复制过程中产生的错误不能及时纠正,并逐渐积累,影响基因表达,导致细胞功能受损;同时,由于无法修复受损的 DNA,参与 DNA损伤反应的因子在衰老细胞中经常被过度激活,持续的 DNA损伤会诱导白细胞介素6(IL- 6)等炎症因子的分泌。此外,过度激活的 DDR,例如双链断裂等严重DNA损伤引起的 DDR,会导致 CHK2的磷酸化和激活,最终导致检查点激活和细胞周期阻滞。
(二)端粒缩短(telomereattrition)
端粒(telomere)是存在于真核细胞染色体末端的一小段DNA- 蛋白质复合体,端粒的短重复序列与端粒结合蛋白一起构成了特殊的染色体末端“帽子”结构,作用是保持染色体的完整性和控制细胞分裂周期。人类出生时,端粒由8~15kb的短串联重复序列(5′- TTAGGG- 3′)组成,由于末端复制问题,端粒在每个复制周期后都会缩短。在早期发育过程中,端粒 DNA可被端粒酶(telomerase)延长,以抵消由于细胞高度增殖而在每个复制周期后缩短的 50~200个核苷酸。然而,在大多数体细胞中,胚胎发育阶段结束时,端粒酶也会失活,端粒只能在增殖过程中逐渐缩短,而当端粒缩短至 Hayflick极限值时,它会诱导永久性DDR,触发不可逆的细胞周期停滞。因此,端粒 DNA长度(telomerelength,TL)不仅是细胞有丝分裂的时钟,而且也是评估个体衰老的时钟。
许多不同物种(包括人类和小鼠在内)的正常衰老过程中都可以观察到端粒缩短。人类端粒酶缺乏与肺纤维化、再生障碍性贫血和先天性角化不良等疾病的过早发展有关,均与阻碍了受影响组织的再生能力不无关系。转基因动物模型已经揭示了端粒磨损、细胞衰老和有机体衰老之间的因果关系。端粒缩短或延长的小鼠的寿命也相应地缩短或延长。当端粒酶被基因重新激活时,端粒酶缺陷小鼠的过早衰老可以恢复。同样,在阿尔茨海默病模型中,维持成年神经元正常水平端粒酶的小鼠细胞存活数目增多,认知功能得以维持。因此,衰老可以通过端粒酶的激活来进行调控。
(三)表观遗传改变(epigeneticalterations)
表观遗传改变是指在核酸序列未发生任何改变的情况下,遗传物质转录异常。衰老中常见的表观遗传改变包括基因组 DNA甲基化改变、组蛋白修饰异常、异常染色质重塑、非编码RNA(ncRNA)功能失调等。大量的酶系统也参与了表观遗传模式的产生和维持,这些酶包括DNA甲基转移酶、组蛋白乙酰化酶、去乙酰化酶、甲基化酶和去甲基化酶,以及与染色质重塑或ncRNA合成和成熟有关的蛋白质复合物。
目前,基于选定位点 DNA甲基化状态的表观遗传时钟(epigeneticclocks)被引入,以预测按时间顺序排列的年龄和死亡率风险,并评估可能延长人类寿命的干预措施。组蛋白的整体缺失及其翻译后修饰的组织赖性变化也与衰老密切相关。组蛋白表达的增加延长了果蝇的寿命,组蛋白去甲基化酶通过靶向胰岛素/胰岛素样生长因子- 1(insulin-likegrowthfactor-1,IGF- 1)信号通路等关键寿命途径的组成部分来调节寿命。其他组蛋白修饰酶,如蛋白质脱乙酰酶和 ADP核糖基转移酶的 SIRT家族成员,也有助于健康衰老。除DNA和组蛋白修饰因子外,一些染色体蛋白和染色质重塑因子,如与基因组稳定性 DNA修复有关的异染色质蛋白 1-α(heterochromatinprotein1-α,HP1-α)和多梳组蛋白(polycombgroupprotein),也可能调节衰老。这些表观遗传因子的改变导致染色质结构的深远变化,包括整体异染色质丢失和重塑,这是衰老细胞中常见的事件。大量且不断增长的非编码 RNA
(non- codingRNA,ncRNA),包括长链非编码 RNA(longnoncodingRNA,lncRNA)、微 RNA(microRNA, miRNA)和环状 RNA,已成为影响衰老的表观遗传因素。功能获得和丧失研究首次证实了几种miRNA具有调节无脊椎动物寿命的能力。
(四)蛋白质稳态失衡(lossofproteostasis)
许多与年龄相关的疾病,如肌萎缩侧索硬化(amyotrophic lateral sclerosis,ALS)、阿尔茨海默病、帕金森病和白内障,其发病机制都与蛋白质稳态失衡有关:多种衰老因素会导致肽段及蛋白质的错误折叠、氧化、糖化或泛素化。由于错误翻译、错误折叠或不完整蛋白质的产生增加,细胞内蛋白质稳态
(proteostasis)被破坏,异常的蛋白质易于形成聚集体,成为细胞内包涵体或细胞外淀粉样斑块,从而导致细胞功能异常,引起疾病。
在实验中,通过各种手段增加蛋白质稳态均可以延缓衰老过程。对核糖体蛋白RPS23进行基因编辑,以提高 RNA到蛋白质翻译的准确性,可以延长庞氏裂殖酵母、秀丽隐杆线虫和黑腹果蝇的寿命;而 RPS9中的某些突变会增加错误翻译的概率,导致小鼠过早衰老。蛋白质的翻译延伸减慢和氧化损伤累积,也会使“分子伴侣”从正确折叠的蛋白质中解离,导致内质网应激。将重组人 HSP70蛋白应用于小鼠可增强蛋白酶体活性,降低脑脂褐素水平,改善认知功能,延长寿命。同样,对衰老小鼠施用化学类分子伴侣4- 苯基丁酸可以降低大脑内质网应激水平并改善认知。
二、细胞层面衰老标志
(一)自噬功能障碍(disabled macroautophagy)
受损细胞器及异常蛋白质的堆积会导致衰老、引起疾病,而巨自噬(macroautophagy),即“自噬”,就是细胞内主要的清除机制。通过形成具有双层膜结构的自噬小体,识别并包裹异常蛋白质、受损或老化的细胞器、异位胞质DNA甚至入侵的病原体,继而与细胞内的溶酶体相结合,由溶酶体酶降解目标。
自噬受到相关基因的调控。人类自噬相关基因如 ATG5、ATG7和 BECN1的表达随着年龄的增长而下降。实验小鼠 ATG5敲低会导致多器官系统的早衰及寿命缩短。自噬流(autophagicflux)的减少会导致蛋白质形成聚集体,导致功能失调细胞器的积累,减少病原体的清除,并引起炎症。另外,长寿个体中的 CD4+T淋巴细胞具有更强的自噬活性,而来自衰老供体的血液 B淋巴细胞和 T淋巴细胞的自噬也会减少。抑制自噬会显著增加恶性肿瘤的发生率,说明自噬与癌症免疫监测相关。调节或执行自噬的基因功能缺失、突变与广泛的心血管疾病、感染性疾病、神经退行性疾病、代谢性疾病、肌肉骨骼疾病、眼部疾病和肺部疾病具有因果关系,其中许多疾病在组织病理和功能水平上类似于过早衰老。
(二)营养感知失调(deregulatednutrient-sensing)
营养感应网络在进化过程中高度保守,它由多种细胞外配体(如胰岛素和 IGFs)、细胞表面受体及细胞内信号级联反应组成。这些级联反应涉及PI3K- Akt和 Ras- MEK- ERK通路,以及包括 FOXO、 TFEB在内的多个转录因子家族。雷帕霉素复合物- 1(MTORC1)可对营养物质(包括葡萄糖和氨基酸)以及应激原(如缺氧和低能量)作出反应,以调节包括转录因子(如 SREBP和 TFEB)在内的多种蛋白质的活性,从而调节多种基因的表达,发挥众多功能。
因此,营养感应网络是细胞活动的中心调节器,调节不同营养状态下的自噬,mRNA和核糖体的生物发生,蛋白质合成,葡萄糖、核苷酸和脂质代谢,线粒体的生物发生和蛋白酶体的活性。细胞随时感知外界营养水平,当营养素充足时,细胞获取营养物质来加速自身的生长和代谢;当营养素缺乏时,细胞通过调节自身代谢水平、激活自噬等分解代谢途径,以达到营养物质的循环利用从而维持其存活的目的。随着年龄的增长,机体细胞会出现对葡萄糖、脂肪、酮等能量底物的识别和反应能力下降的现象,该现象被称为营养感知失调。
在多种动物模型中,营养感应网络组成部分的活性降低可以延长寿命。其中,将雷帕霉素抑制剂 mTOR用于抗衰老治疗,就是一大突破。mTOR信号通路感受并整合细胞内外各种复杂的能量、营养环境信号,并在调节细胞的生长、分化、衰老等过程中发挥重要作用。一方面,抑制mTOR信号可以降低 mRNA整体翻译水平,减少蛋白合成负担和细胞能量消耗,以应对应激环境下的细胞能量危机;另一方面,mTOR信号的下调可以增强原本随衰老降低的自噬能力,以清除细胞内受损蛋白质和细胞器,重建蛋白稳态。
另一种降低营养感知的方法是限制能量摄入,然而,饮食限制方案并不能延长所有小鼠品系的寿命,说明此方案的应用仍须考虑遗传背景。人类饮食限制的效果因临床试验从性差而难以得出结论,但可明确其对免疫和炎症有积极影响。改良后的间歇性禁食方案可以避免热量限制引起的小鼠长期体重减轻,从而延长其寿命。
(三)线粒体功能紊乱(mitochondrialdysfunction)
线粒体是重要的胞内细胞器,在能量供应、钙稳态、凋亡调节、呼吸链复合物合成等多种细胞活动中发挥重要作用。衰老细胞的线粒体动力学及形态会发生异常改变,如线粒体嵴断裂,线粒体肿胀、长度增加,线粒体融合/分裂比例失调。线粒体动力学失衡会导致线粒体稳态失衡及其功能障碍。衰老细胞的线粒体生物合成途径相关分子表达及活性降低,能量生成减少,活性氧(reactiveoxygen species,ROS)产生增多,并可能引发线粒体膜通透性增高,导致炎症和细胞死亡。
线粒体具有自己独特的环状线粒体 DNA(mitochondrialDNA,mtDNA),可编码自身呼吸链,以及多个小的调节肽,这些肽段统称为线粒体衍生肽(mitochondrial- derivedpeptide,MDP)。当 mtDNA突变产生缺陷时,氧化呼吸链的缺失会导致过氧化氢、超氧阴离子、羟自由基等多种 ROS过量产生,而增龄条件下自由基清除系统能力不断衰退,导致体内自由基累积及氧化应激的发生,引起蛋白质、脂质、DNA等大分子损伤,破坏mtDNA和线粒体膜稳定性,使受损线粒体产生更多 ROS,进一步加剧线粒体的功能障碍,引起细胞衰老;另外,ROS还可对端粒造成损伤,导致端粒缩短,加速细胞的衰老。
humanin是最早发现的一种MDP,在进化过程中高度保守,并在生物体内广泛存在。研究发现,humanin的血浆水平随着年龄的增长而下降,但是百岁老人和他们的后代均表现出高水平的humanin。另一种新发现的 MDP是线粒体 12SrRNA- c 的开放阅读框(mitochondrialopenreading frameofthe12SrRNA- c,MOTS- c),其血浆水平也随着年龄的增长而下降,但可以通过运动诱导。MOTS- c有利于产生内源性 AMPK激动剂 AICAR,从而预防高龄和高脂肪饮食诱导的肥胖及胰岛素抵抗。因此,MDP成为了一类潜在的抗衰老因子。
其他积极提升线粒体功能的手段也可以延长寿命。例如,左旋肉碱会随年龄增长而降低,而补充左旋肉碱对体弱的受试者和老年男性都有积极影响,这种积极作用可能是由于左旋肉碱可以限制线粒体对脂肪酸的氧化。
(四)细胞衰老(cellularsenescence)
细胞衰老指原本具有分裂能力的细胞逐渐失去分裂增殖能力,进入持续的、不可逆的分裂停滞状态。细胞衰老最明确的标志之一是细胞周期稳定地停滞在G1期或 G2期。各种衰老应激原,如端粒缩短、DNA损伤、氧化应激、化疗药物、紫外线和辐射等都可引起细胞的 DNA 损伤,并激活 p16/pRB信号和/或 p53/p21信号导致细胞周期阻滞,以抑制损伤细胞增殖。
在衰老过程中,与衰老相关的系统和细胞间信号中断,加上遗传物质损伤和线粒体功能障碍,导致癌细胞和衰老细胞出现程序性细胞死亡(programmedcelldeath,PCD)抵抗。同时,衰老细胞异质表达的免疫检查点蛋白程序性死亡受体配体1(PD- L1),具有免疫细胞抗性,从而协助衰老细胞逃避免疫系统的清除,使得衰老细胞堆积,导致各种年龄相关疾病。
除细胞增殖受阻、细胞凋亡耐受及免疫细胞抗性外,细胞衰老的第四个特征为衰老相关分泌表 型(senescence- associatedsecretoryphenotype,SASP),指 衰老细胞合成和分泌过多的可溶性因子,包括促炎性细胞因子、趋化因子、血管生成因子、生长调节剂和基质金属蛋白酶(matrix metalloproteinase,MMP)。SASP是衰老细胞的典型标志,会以自分泌和旁分泌的方式加速衰老,形成衰老微环境,导致炎症反应。在机体发育和成熟期,炎症作为抵御有害物质入侵的防御机制,通过免疫细胞的激活消除病原体和促进组织修复来保护宿主免受侵害;但随着年龄不断增长,免疫系统功能在老龄时逐渐衰退,难以消除衰老细胞持续分泌SASP造成的炎症环境,使机体长期处于慢性炎症状态。
三、整体层面衰老标志
(一)干细胞耗竭(stemcellexhaustion)
干细胞对于维持组织稳态和再生至关重要。衰老时组织更新减少,损伤后的组织修复功能也受损。在增龄过程中,干细胞数量和功能进行性下降,称为干细胞耗竭。如前所述,在衰老过程中,干细胞也会积累 DNA损伤,经历表观遗传学变化、自噬与代谢失调等,导致干细胞本身的功能障碍和衰竭。同时,随着组织细胞不断地衰老与清除,需要动员干细胞以进行组织再生。以造血干细胞
(hematopoieticstem cell,HSC)为例,大多数造血干细胞很少进行周期性自我更新或分化为子代的细胞。随着每一次细胞分裂,HSC分化为血细胞的潜能下降,因而需要大量的造血祖细胞以弥补单个细胞功能的下调。HSC自身的大量增殖会导致 HSC的耗竭。
另外,衰老的组织细胞分泌的 SASP也会诱导干细胞的衰老,在三重作用之下,干细胞的衰老与耗竭最终会导致组织再生能力受损,影响器官功能,诱发生物体衰老。
(二)细胞间通信改变(alterationsinintercellularcommunication)
细胞间通信涉及神经内分泌因子和多种激素相关的信号通路,如肾上腺素、多巴胺和胰岛
素/IGF1通路。尽管这些分子信号通路的变化往往来自细胞内部蛋白或基因的表达异常,但这些分子信号通路之间的对话及其在整个机体中的信息传递功能,会把一个细胞的异常放大为局部组织甚至整个机体的炎症反应及免疫响应。因此,细胞间信号通路的改变会影响整个机体的基因组和微生态稳定。
血液中的促衰老因子(如 CCL11)可使神经细胞前体细胞减少,IL-6和 TGF-β会损害造血干细胞,而 C1q则损害肌肉修复,理论上清除这些因子可能有抗衰老效应。血液中的抗衰老因子(如 CCL3)可以重新活化造血干细胞,TIMP2重新活化海马体,IL- 37改善运动耐力,GDF11则可重新活化肌肉、脑组织等。细胞间通信还包括神经系统对脏器的远程控制,以及短程交互(如 ROS 和细胞因子)。
衰老还伴有细胞外基质的损伤,如 AGE、细胞外酶的积聚导致纤维化。基质坚硬度增加也影响肌细胞功能。抑制 PIEZO1通路、YAP/ TAZ或修复Ⅰ型胶原蛋白可帮助抗衰老。总体来看,细胞间信号通路和细胞外基质的变化与衰老密切相关。
(三)慢性炎症(chronicinflammation)
炎症在衰老过程中增加,并伴有全身症状及局部病理表型,包括动脉硬化、神经炎症、骨关节炎和椎间盘退变,因此也称为炎性衰老。炎症细胞因子和生物标志物[如 C反应蛋白(C- reactiveprotein, CRP)]在血液循环中的浓度随着年龄的增长而增加。血浆中 IL- 6水平升高是老年人全因死亡率的预测性生物标志物。
与衰老炎症增强相关的是免疫功能下降。衰老时 T细胞群的变化导致促炎性TH1和 TH17细胞功能亢进。免疫监测缺陷导致难以消除病原体,以及恶性或衰老的细胞。与自我耐受性的丧失相伴而来的,是与年龄相关的自身免疫性疾病的增加,以及生物屏障的维护和修复减少,以上变化均会促进全身炎症的发生。研究发现,抑制TNF-α、IFN-γ受体、PGE2受体或细胞因子(如 IL- 1β),可以改善肌少症及认知障碍,延长寿命。临床试验表明IL- 1β抑制剂可改善糖尿病和癌症。非甾体抗炎药(如阿司匹林)也可能延长寿命,但需要更大样本研究。总体来说,控制炎症水平对延长寿命非常重要。
炎性衰老也是一系列促衰老机制共同作用的结果:如 DNA损伤、表观遗传失调、蛋白质稳态失衡、生长激素- 胰岛素轴激活、自噬降低、线粒体功能降低以及免疫能力降低。
(四)菌群生态失调(dysbiosis)
肠道微生物组参与营养物质的消化、吸收以及必需代谢产物的产生,包括维生素、氨基酸衍生物、次级胆汁酸和短链脂肪酸(short- chainfattyacid,SCFA)。肠道微生物菌群还向外周和中枢神经系统以及其他远处器官发出信号,并对宿主健康的整体维持产生强烈影响。这种细菌- 宿主双向通信的破坏可导致生态失调,并引起多种病理状况,如肥胖、2型糖尿病、溃疡性结肠炎、神经系统疾病、心血管疾病和癌症。
随着年龄的增长,个体呈现出肠道微生物组的特异性,衰老可能存在多种肠道微生物组的轨迹。病理性衰老的多组学研究表明,两种不同的早衰症小鼠模型均表现出肠道微生态失调。粪便微生物群移植揭示了肠道微生态失调在慢性全身炎症及衰老相关适应性免疫下降中的致病作用。肠道微生物群从老年小鼠转移到年轻无菌小鼠体内可引发炎症反应,其特征是脾脏中 CD4+T细胞分化增强、炎症细胞因子上调。总之,肠道微生物菌群的数量、结构及代谢功能等方面的变化很可能会促进机体衰老的进程,这将是人体衰老研究中的一个重要方向。
综上所述,目前明确的 12个衰老标志体现了衰老时分子层面的基因结构、修饰,到转录翻译、蛋白表达变化,再到细胞层面的不同蛋白质之间、细胞器之间的相互影响,最后到整体层面的细胞之间、系统之间的相互作用、相互影响,形成了整体衰老。各系统衰老细胞的逐渐堆积,影响到组织、器官、系统的功能变化,促进了衰老的发生发展进程(图 5- 3)。
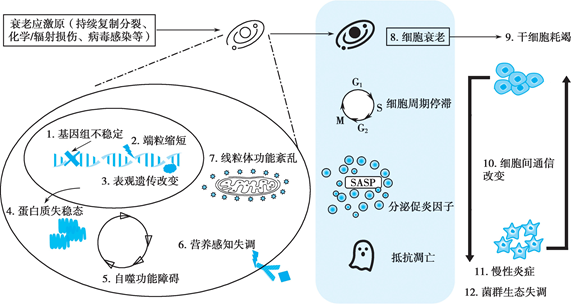
图 5- 3 衰老的发生发展机制及联系

