生理条件下,心排血量可以随着机体代谢需要的升高而增加,这主要是通过对心率、心室前、后负荷和心肌收缩性的调控实现的。
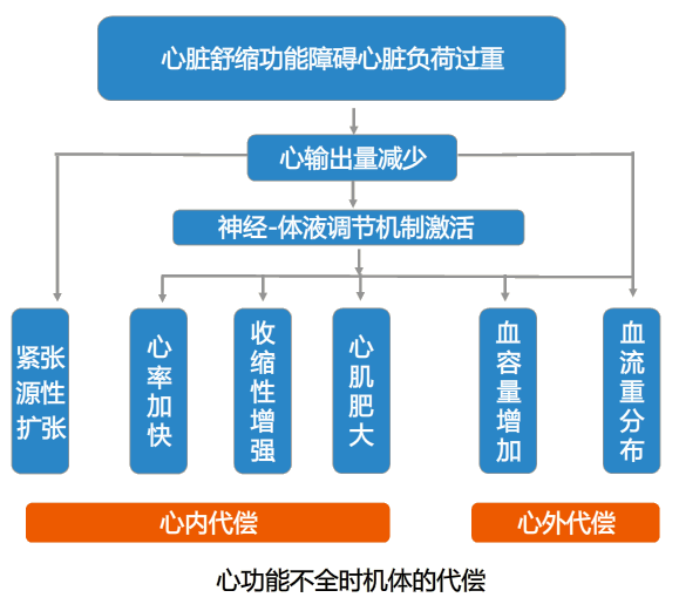
心脏泵血功能受损时,心排血量减少可以通过多种途径,引起内源性神经-体液调节机制激活,这是心功能减退时介导心内与心外代偿与适应反应的基本机制,也是导致心功能不全发生与发展的关键途径。
一、神经-体液调节机制激活
神经-体液调节机制激活是心功能减退时介导心内与心外代偿与适应反应的基本机制,也是导致心功能不全发生与发展的关键途径
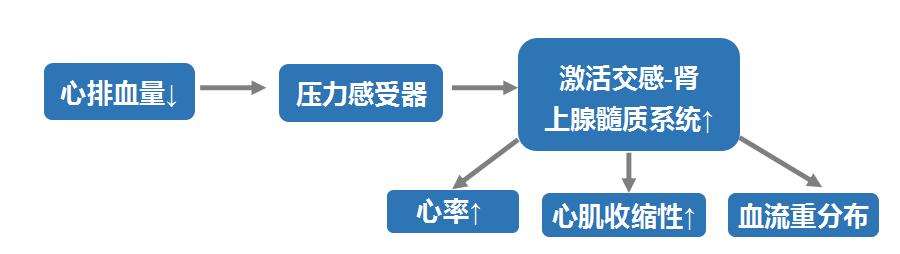
1. 交感-肾上腺髓质系统激活
心功能不全时交感-肾上腺髓质系统激活的作用:
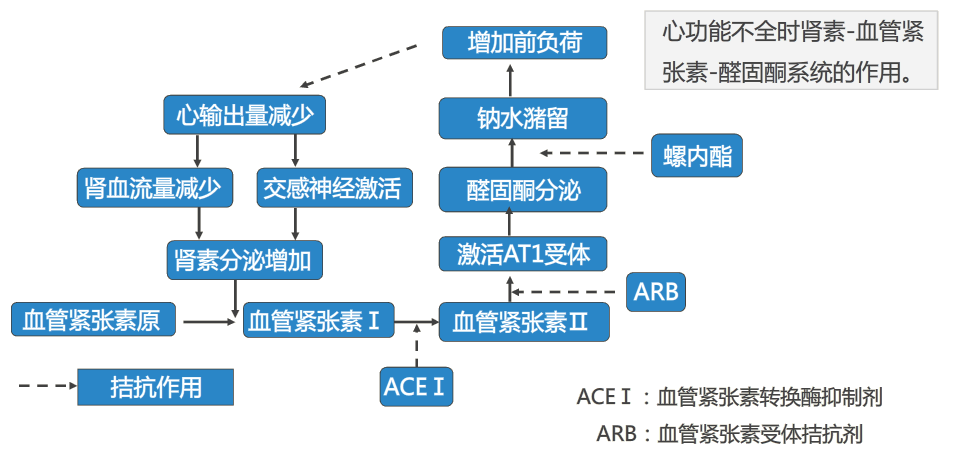
2. 肾素-血管紧张素-醛固酮系统激活
肾脏低灌流、交感神经系统兴奋和低钠血症等都可以激活肾素-血管紧张素醛固酮系统。
AngⅡ增加可以通过直接的缩血管作用及与去甲肾上腺素的协同作用对血流动力学稳态产生明显影响。AngⅡ可以升高肾灌注压,通过肾内血流的重分布维持肾小球血流量,从而维持肾小球滤过率。醛固酮增加可引起钠潴留,通过维持循环血量保持心排血量正常。
3. 钠尿肽系统激活
心房肌:心房钠尿肽(ANP)
心室肌:B型钠尿肽(BNP)
N末端B型钠尿肽原(NT-proBNP):无生物学活性
功能:利钠排尿,扩张血管和抑制肾素及醛固酮的作用
血浆BNP/NT-proBNP:心功能不全诊断和鉴别诊断、风险分层以及评估预后
在神经-体液机制的调控下,机体对心功能降低的代偿反应可以分为心脏本身的代偿和心外代偿两部分。
二、心脏本身的代偿
心脏本身的代偿形式包括心率增快、心脏紧张源性扩张、心肌收缩性增强和心室重塑(ventricular remodeling)。
其中,心率加快、心脏紧张源性扩张和心肌收缩性增强属于功能性调整,可以在短时间内被动员起来;
而心室重塑是心室在前负荷和后负荷长期增加时,通过改变心室的结构、代谢和功能;而发生的慢性综合性代偿适应性反应。
1. 心率增快
心排血量是每搏输出量与心率的乘积,在一定的范围内,心率加快可提高心排血量,并可提高舒张压,有利于冠脉的血液灌流,对维持动脉血压、保证重要器官的血流供应有积极意义。
心功能损伤时,心率加快的机制主要是:
①压力感受器的调控:由于心排血量减少,主动脉弓和颈动脉窦压力感受器的刺激碱弱,经窦神经传到中枢的抑制性冲动减少,交感神经兴奋,引起心率加快;
②容量感受器的调控:心脏泵血减少使心腔内剩余血量增加,心腔舒张末期容积和压力升高,可刺激位于心房和心室的容量感受器经迷走神经传入纤维至中枢,使迷走神经抑制、交感神经兴奋;
③化学感受器的调控:如果合并缺氧,可以刺激主动脉体和颈动脉体化学感受器,反射性引起心率加快。
此外,焦虑、恐惧、应激、创伤和发热等刺激也可激活交感神经。
心率加快是一种易被快速动员起来的代偿反应,往往贯穿于心功能不全发生和发展的全过程。
心率加快的代偿作用也有一定的局限性,其原因是:
①心率加快增加心肌耗氧量;
②心率过快(成人>180次/min)明显缩短心脏舒张期,不但减少冠脉灌流量,使心肌缺血、缺氧加重,而且缩短心室充盈时间,减少充盈量,心排血量反而降低。
2. 心脏紧张源性扩张
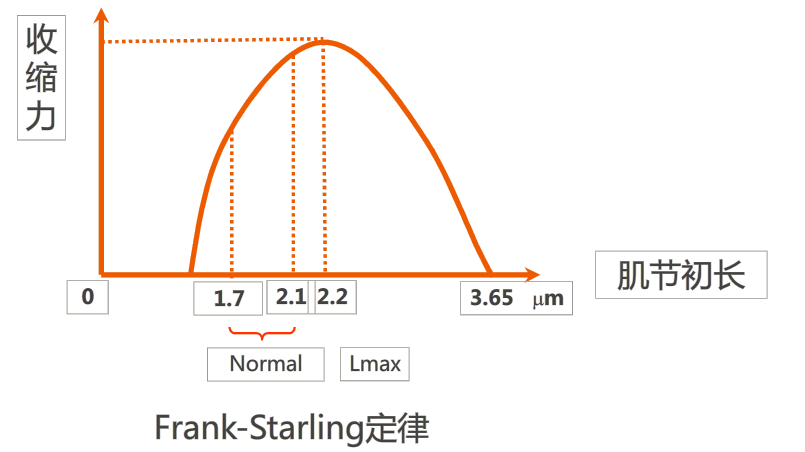
当心脏收缩功能受损时,心脏本身会发生快速的、应急性的调节反应。
由于每搏出量降低,使心室舒张末期容积增加,前负荷增加导致心肌纤维初长度增大(肌节长度不超过2.2μm),此时心肌收缩力增强,代偿性增加每搏输出量,这种伴有心肌收缩力增强的心腔扩大称为心脏紧张源性扩张,有利于将心室内过多的血液及时泵出。
但长期前负荷过重引起的心力衰竭以及扩张性心肌病主要是引起肌节过度拉长,使心腔明显扩大。这种心肌过度拉长并伴有心肌收缩力减弱.的心腔扩大称为肌源性扩张,其已失去增加心肌收缩力的代偿意义。
3. 心肌收缩性增强
在心功能损伤的急性期,心肌收缩性的增强对于维持心排血量和血流动力学稳态是十分必要的代偿和适应机制。
而慢性心功能不全时,心肌β肾上腺素受体减敏,血浆中虽存在大量儿茶酚胺,但正性变力作用的效果显著减弱。
4. 心室重塑
心脏由心肌细胞、非心肌细胞(包括成纤维细胞、血管平滑肌细胞、内皮细胞等)及细胞外基质(extracellular matrix)组成。
心室重塑是心肌损伤或负荷增加时,通过改变心室的结构、代谢和功能而发生的慢性综合性代偿适应性反应。
(1)心肌细胞重塑
1)心肌细胞肥大
心肌肥大是指心肌细胞体积增大:在细胞水平上表现为细胞直径增宽,长度增加;在器官水平表现为心室质(重)量增加,心室壁增厚。
按照超负荷原因和心肌反应形式的不同又可将超负荷性心肌肥大分为:
向心性肥大
离心性肥大
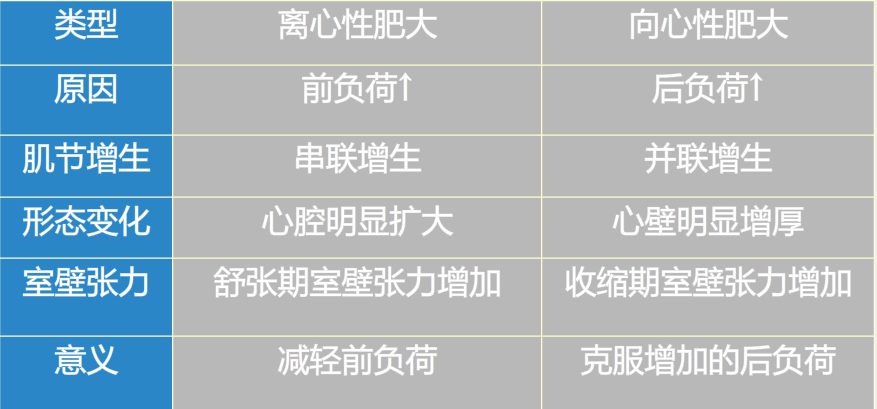
无论是向心性肥大还是离心性肥大都是对室壁应力增加产生的适应性变化,是慢性心功能不时极为重要的代偿方式。
心肌肥大的意义:
作用缓慢、持久。
心肌总收缩力增强,有利于维持心排血量。
室壁厚度增加,降低室壁张力,减少心肌耗氧量。
向心性肥大的代偿能力强于离心性肥大,但二者均可增加心脏作功和心输出量,使心功能在相当长的时间内处于稳定状态,不发生心力衰竭。
代偿作用局限:
肥大心肌的生长具有不平衡性,因此当心肌过度肥大超过某种限度时,则发生由代偿向衰竭的转化。
2)心肌细胞表型的改变
指由于心肌所合成的蛋白质的种类变化所引起的心肌细胞“质”的改变。
胎儿期基因激活: ANP、BNP、β-肌球蛋白重链( B-MHC )等。
某些功能基因的表达抑制:钙泵、离子通道等。
(2)非心肌细胞及细胞外基质的变化
细胞外基质增加,心肌纤维化。
三、心脏以外的代偿
1. 增加血容量
机制:①交感神经兴奋;②RAA系统激活;③ADH增多;④抑制钠水重吸收的激素减少
2. 血流重新分布
皮肤、骨骼肌与内脏器官的血流量减少,心、脑血流量不变或略增加
综上所述,心功能不全时,在神经-体液机制的调节下,机体可以动员心脏本身和心脏以外的多种代偿机制进行代偿,并且这种代偿贯穿于心功能不全的全过程。