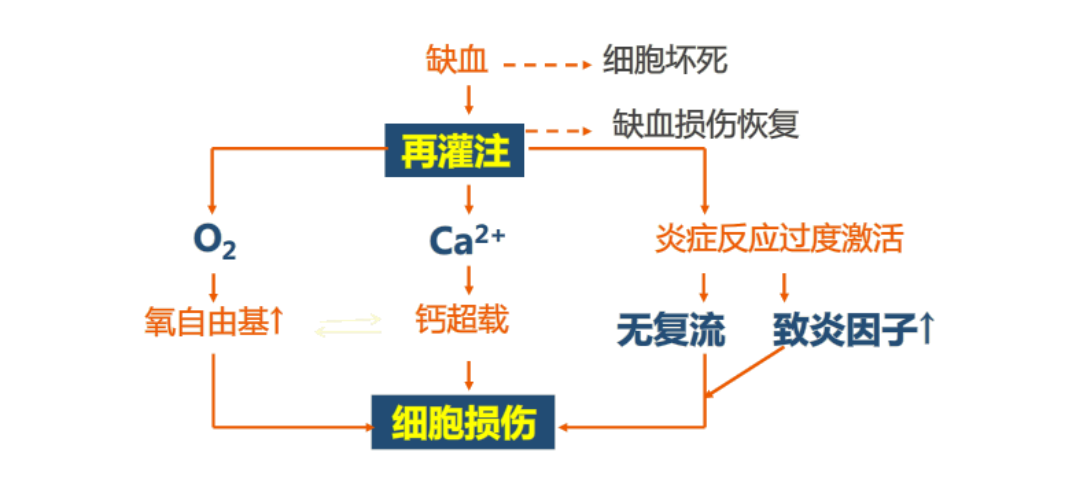
缺血-再灌注损伤的发生机制尚未彻底阐明,目前认为自由基生成增多、细胞内钙超载和炎症反应过度激活是缺血-再灌注损伤的重要发病机制。
一、自由基增多
1. 自由基的概念及分类
自由基(free radical) 是指在外层电子轨道上具有单个不配对电子的原子原子团或分子。

自由基的特点:
化学性质非常活泼,易夺取其他物质的一个电子;氧化性强,半衰期短。
2. 自由基的生成与清除
(1)生成
①氧化磷酸化过程中单电子还原
②其他反应中生成
体内的许多酶促反应和非酶促反应可通过单电子转移而产生自由基。
(2)清除
①抗氧化物质
辅酶Q、维生素E、β-胡萝卜素、维生素C、谷胱甘肽等提供电子使自由基还原,清除自由基
②抗氧化酶
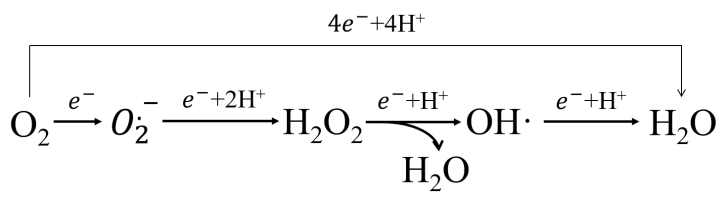
超氧化物歧化酶(superoxide dismutase,SOD)可歧化![]() 生成H2O2
生成H2O2
过氧化氢酶( catalase )可清除H2O2
谷胱甘肽过氧化物酶( glutathione peroxidase , GSH-Px)可清除OH·
3. 缺血-再灌注导致自由基增多的机制
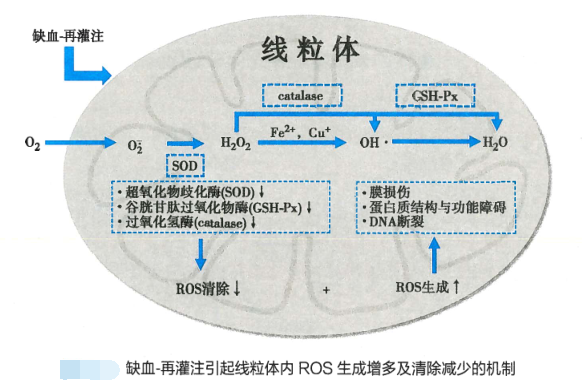
(1)线粒体损伤
线粒体是细胞氧化磷酸化反应的主要场所,当缺血缺氧时细胞内氧分压降低、线粒体氧化磷酸化功能障碍,ATP生成减少,Ca2+进入线粒体增多,细胞色素氧化酶系统功能失调,电子传递链受损,SOD、catalase、GSH-Px等抗氧化酶类活性下降,以致再灌阶段进入细胞内的氧经单电子还原而形成的活性氧增多,特别是线粒体内H2O2及OH·生成增多。
(2)中性粒细胞聚集及激活
中性粒细胞(neutrophils)在吞噬活动时耗氧量显著增加,所摄取的氧绝大部分经细胞内NADPH氧化酶和NADH氧化酶的催化,接受电子形成氧自由基,用以杀灭病原微生物。
缺血时,大量中性粒细胞聚集并激活;
再灌注期间,组织重新获得氧,激活的中性粒细胞耗氧量显著增加,产生大量的氧自由基,即呼吸爆发或氧爆发,进一步造成组织细胞的损伤。
(3)黄嘌呤氧化酶形成增多
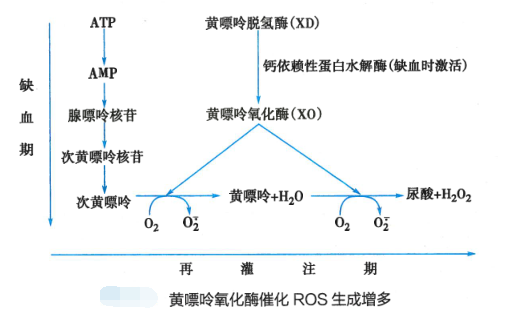
正常情况下黄嘌呤氧化酶(xanthine oxidase,XO)占10%,其前身黄嘌呤脱氢酶(xanthine dehydrogenase, XD)占90%,这两种酶主要存在于毛细血管内皮细胞内。
一方面,缺血组织ATP生成减少,并相继分解为ADP、AMP、腺嘌呤核苷(adenine nucleoside)、次黄嘌呤核苷(hypoxanthine nucleoside)和次黄嘌呤(hypoxanthine), 次黄嘌呤自身不能代谢生成黄嘌呤(xanthine),因此在缺血组织内大量积聚。
另一方面,ATP缺乏使钙泵功能发生障碍,造成细胞内Ca2+大量积聚,激活细胞内Ca2+依赖性蛋白水解酶,促使黄嘌呤脱氢酶大量转化为黄嘌呤氧化酶。
再灌注时,大量分子氧随血液进入缺血组织,在黄嘌呤氧化酶催化次黄嘌呤生成黄嘌呤,并进而催化黄嘌呤转变为尿酸(uric acid)的两步反应中,都以分子氧为电子接受体,生成大量的O2-和H2O2。
H2O2在Fe2+参与下可形成化学性质更为活泼的OH·。
上述反应在再灌注早期尤为强烈,产生大量活性氧。
(4)儿茶酚胺自氧化增强
缺血-再灌注也是一种应激反应,交感-肾上腺髓质系统兴奋产生大量儿茶酚胺。
一方面具有代偿调节作用;
另一方面,通过自氧化可产生大量的氧自由基。
4. 自由基增多引起机体损伤的机制
自由基性质极为活泼,可与其他物质反应,甚至相互反应形成二聚体或多聚体。自由基可破坏多糖,氧化蛋白质,使不饱和脂肪酸过氧化,造成细胞结构功能障碍,甚至水解。
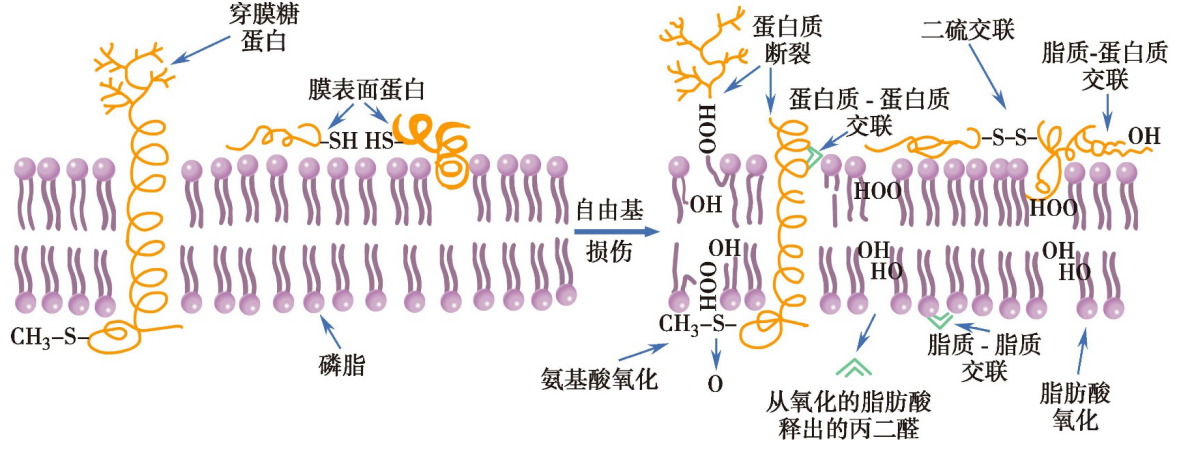
(1)膜脂质过氧化
自由基与不饱和脂肪酸作用引发脂质过氧化( lipid peroxidation)反应,使膜结构受损、功能障碍,引起以下损伤:
细胞及细胞器膜结构破坏
生物活性物质生成增多
ATP生成减少
(2)蛋白质功能抑制
自由基与活性氧可与细胞结构蛋白和酶的巯基氧化形成二硫键,使氨基酸残基氧化,胞质及膜蛋白和某些酶交联形成二聚体或更大的聚合物,直接损伤蛋白质的功能,如离子通道蛋白或转运体功能抑制。
同时膜磷脂微环境的改变共同导致跨膜离子梯度异常,Na+、Ca2+内流,细胞肿胀与Ca2+超载。
脂质过氧化可抑制膜受体、G蛋白与效应器的耦联,引起细胞信号转导功能障碍。
(3)核酸破坏与DNA断裂
自由基可使核酸碱基羟化及DNA断裂,这种作用80%为OH·所致。
总之,缺血-再灌注会使自由基生成增多,特别是氧自由基与活性氧,从而加重细胞损伤。
由于氧化物质增多而抗氧化防御机制降低之间的不平衡导致的损伤,又被称为“氧化应激"。
二、钙超载
生理情况下,细胞内游离Ca2+浓度约为0.1µmol/L,细胞外游离Ca2+浓度约为1.0mmol/L, 细胞膜内外Ca2+浓度相差1万倍。
细胞通过转运机制维持细胞内外钙浓度差,保持细胞内低钙的状态,称为钙稳态。
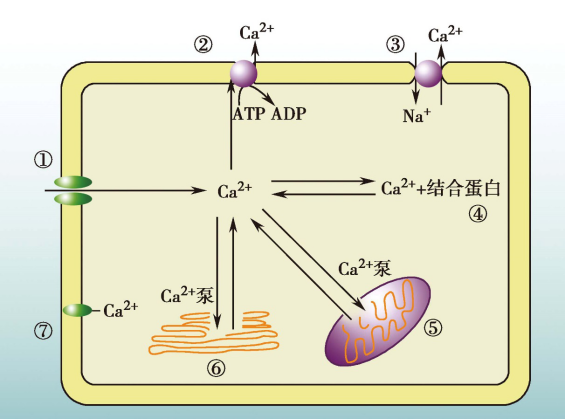
细胞Ca2+转运模式图
当各种原因引起细胞Ca2+转运机制异常、细胞内Ca2+含量增多,导致细胞结构损伤和功能代谢障碍,称为钙超载( calcium overload) 。
1. 缺血-再灌注导致钙超载的机制
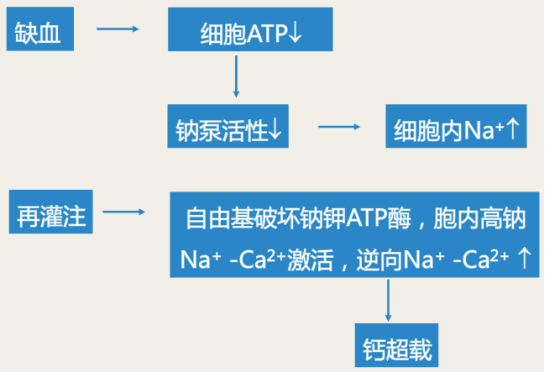
(1)Na+-Ca2+交换异常
Na+/Ca2+交换蛋白的反向运转增强是导致缺血-再灌注时Ca2+超载的主要途径.
直接激活
间接激活
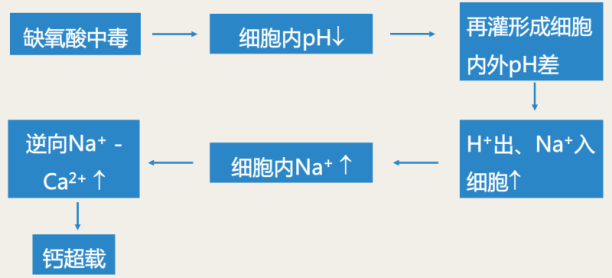
(2)蛋白激酶C(PKC)激活
组织缺血时,儿茶酚胺(CA)释放增加,作用于α1和β肾上腺素能受体,可以间接的激活钠钙交换蛋白。
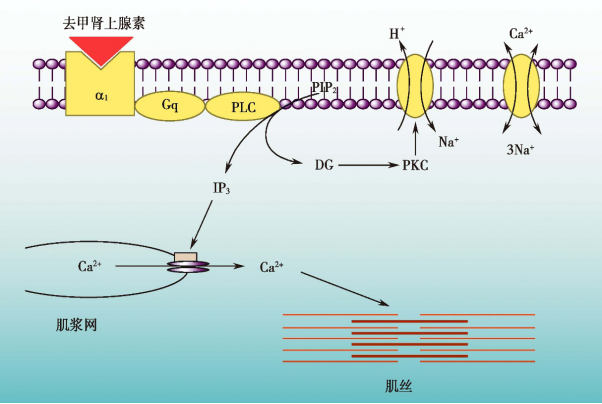
(3)生物膜损伤
生物膜包括细胞膜及细胞器膜,其结构与功能完整是维持细胞内外离子平衡的重要保证。
缺血时由于:①细胞膜对Ca2+的通透性增大,因此Ca2+内流增加;②ATP产生减少,细胞膜及肌浆网钙泵功能低下而不能充分排出或储存Ca2+,亦导致胞浆内Ca2+增多。缺血-再灌注过程中生成的大量氧自由基,通过引发膜的脂质过氧化,导致膜结构受损,一方面细胞膜对Ca2+的通透性增高,细胞外Ca2+内流增多;另一方面肌浆网通透性增高,贮存的Ca2+释出增多,以上均促成钙超载的发生。
2. 钙超载引起机体损伤的机制
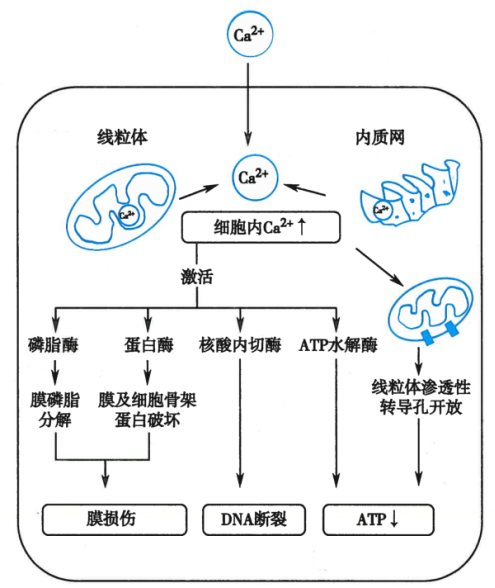
(1)能量代谢障碍
聚集于胞质内Ca2+被线粒体摄取时可消耗大量ATP,同时进入线粒体的Ca2+与含磷酸根的化合物结合,形成不溶性磷酸钙,既干扰线粒体的氧化磷酸化,使ATP生成减少,又损伤线粒体膜而加重细胞能量代谢障碍。
(2)细胞膜及结构蛋白分解
细胞内Ca2+增加可激活磷脂酶类,促使膜磷脂降解,造成细胞膜结构受损。
还可激活钙依赖性蛋白酶活性,促进细胞膜和结构蛋白的分解,激活核酸内切酶,引起染色体的损伤。
(3)加重酸中毒
细胞能量代谢障碍,有氧氧化生成ATP减少,无氧酵解增强,乳酸增多,细胞酸中毒;
细胞内Ca2+浓度升高可激活某些ATP酶,导致细胞高能磷酸盐水解,释放出大量H+,加重细胞内酸中毒。
综上所述,钙超载既是缺血-再灌注的结果,,又是缺血-再灌注细胞损伤的原因。
细胞内Ca2+聚积不仅激活磷脂酶,使膜磷脂降解,又进-步增加细胞膜对Ca2+的通透性,促进钙超载。
三、炎症反应过度激活
1. 缺血-再灌注引起炎症反应过度激活的机制
缺血-再灌注时,白细胞(主要是中性粒细胞)明显增加。
组织缺血-再灌注时白细胞浸润增加的可能机制:
(1)黏附分子生成增多
中性粒细胞激活可使细胞黏附分子表达增加。
(2)趋化因子与细胞因子生成增多
再灌注时,细胞膜磷脂降解,花生四烯酸其代谢产物增多,其中白三烯、血小板活化因子、补体、激肽等具有很强的趋化活性,能吸引大量中性粒细胞进入缺血组织。中性粒细胞自身也能合成、释放多种具有趋化作用的炎症介质。
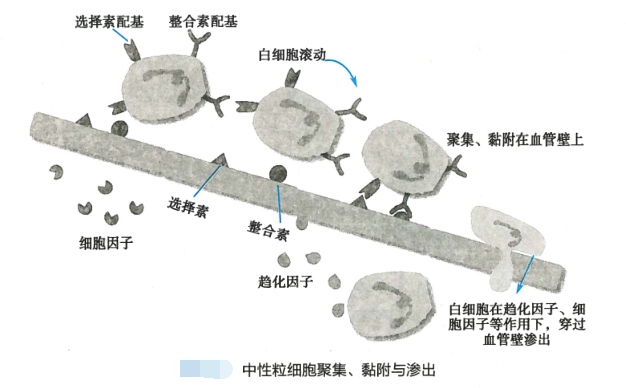
2. 炎症反应引起机体损伤的机制
(1)微血管损伤
①微血管血液流变学改变
正常情况下,血细胞位于血管中心流动,与血管内皮细胞基本不接触,以保证血液的高速流动。
缺血-再灌损伤可引起大量中性粒细胞聚集、黏附在血管内皮细胞上,而且不易分离,极易嵌顿、堵塞微循环血管;加之内皮细胞肿胀、血小板黏附、微血栓形成和组织水肿等,更易形成无复流(no-reflow)现象,加重组织缺血缺氧。
无复流现象:恢复血流灌注后,缺血区依然得不到充分的血液灌流的现象。
②微血管通透性增高
(2)细胞损伤
激活的中性粒细胞与血管内皮细胞可释放大量的活性物质
白细胞浸润等炎症反应进一步过度激活
总结:
缺血-再灌注损伤发生的基本机制,主要是缺血再灌注的过程中自由基生成增多、钙超载及炎症反应过度激活,三者相互作用、协同作用,最终引起细胞、机体损伤。