
细胞信号转导障碍、增强均会导致细胞功能代谢的紊乱而引起疾病或促进疾病的发生发展。
一、家族性肾性尿崩症
概念
中枢性尿崩症;因抗利尿激素(antidiuretic hormone,ADH)分泌减少引起的以多饮和多尿为特征的综合征称为中枢性尿崩症;
家族性肾性尿崩症( familial nephrogenic diabetes insipidus , FNDI):系遗传性肾集合小管上皮细胞膜上的Ⅰ型抗利尿激素(ADH)受体(V2R)数目减少或功能缺陷,使其对ADH的反应性降低,导致对水的重吸收减弱而引起的尿崩症。
发病机制
大多数家族性肾性尿崩症的发病是由于编码V2受体的基因突变造成的:
①突变导致V2受体mRNA的转录受阻,使合成的受体蛋白减少;②突变导致蛋白质的翻译异常,合成的受体缺乏转位的信号肽或不能糖基化,滞留在内质网或高尔基体中,不能转移到细胞膜表面;③突变影响了V2受体的配体结合区,使受体与ADH的亲和力降低;④突变干扰了V2受体与Gs蛋白的偶联,减少对腺苷酸环化酶的激活。总之,不论是V2受体的数量减少或结构异常,最终导致远端肾小管和集合管上皮细胞上的V2受体对外源性和内源性ADH反应性降低,cAMP生成减少,对水的重吸收降低。
少部分家族性肾性尿崩症是由于ADH受体后缺陷(postreceptor defect),其发病机制是位于染色体12q13的编码AQP2的基因突变,使AQP2在细胞内的穿梭功能障碍,使肾小管对水的重吸收减少。
疾病特征及临床表现
NDI患者躲在1岁以内发病,男性显示症状,女性携带者一般无症状。
临床表现:口渴、多饮、多尿等,但血中ADH水平在正常水平以上。
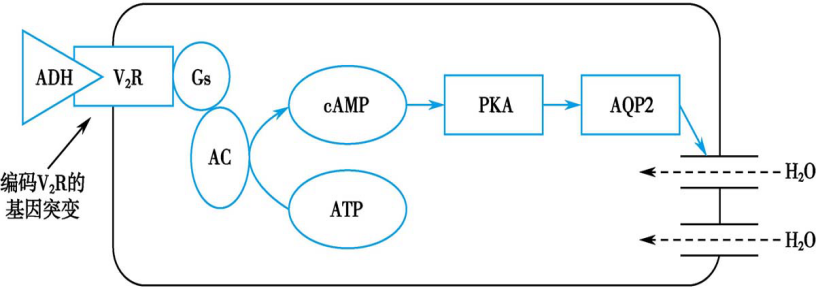
图10-8 家族性肾性尿崩症的发生机制示意图
二、肢端肥大症和巨人症
因垂体生长激素腺瘤使生长激素分泌过多引起的内分泌紊乱性疾病,肢端肥大症和巨人症是其特征性表现。
GH分泌的调节
正常情况下,生长激素由腺垂体合成和释放,受下丘脑分泌的生长激素释放激素(growth hormone releasing hormone)和生长抑素(somatostatin)的双重调节。生长激素释放激素与腺垂体细胞膜上的生长激素释放激素受体结合而激活Gs,导致腺苷酸环化酶活性升高和cAMP增加,进而促进腺垂体细胞合成和分泌生长激素。生长抑素则通过与腺垂体的生长抑素受体结合而激活Gi,减少cAMP水平,抑制生长激素的分泌。
发病机制
在分泌生长激素过多的垂体腺瘤中,约有30%~40%的患者是由于编码Gsα的生长刺激蛋白(gsp)基因发生点突变,最常见的突变位点是精氨酸201或谷氨酰胺227为其它氨基酸残基取代,抑制了G蛋白的GTP酶活性,不能水解GTP,使Gsα处于持续激活状态,cAMP含量增加,促进垂体细胞生长和增加生长激素分泌。
亦有部分垂体腺瘤患者的信号转导障碍机制是垂体肿瘤细胞生长抑素受体后的Gi功能缺陷,对腺苷酸环化酶的抑制作用减弱,从而导致生长激素的过度分泌。
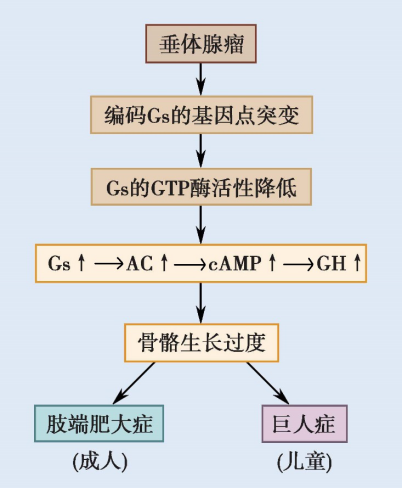
图10-9 肢端肥大症和巨人症的发生机制示意图
对机体的影响
生长激素的功能是促进细胞生长和物质代谢。肢端肥大症或巨人症起病隐匿,呈慢性进展性过程。长期生长激素的过度分泌可刺激骨和软骨过度增生,在儿童引起巨人症,在成人引起肢端肥大症。过量生长激素还可导致全身软组织增生,引起面容改变、鼻大唇厚、皮肤粗厚等。此外,糖尿病、高血压及心脑血管疾病、代谢紊乱性疾病的发生率也会相应增加。
三、恶性肿瘤
正常细胞的增殖、分化及凋亡受到精细的网络调节,细胞癌变最基本的特征是增殖失控分化障碍及凋亡异常。近年来人们认识到绝大多数的癌基因表达产物都是细胞信号转导系统中的重要分子,调控细胞的生存和死亡,从多个环节干扰细胞信号转导过程,导致细胞过度增殖、异常分化和调亡减少,从而导致恶性肿瘤发生(图10-10)。
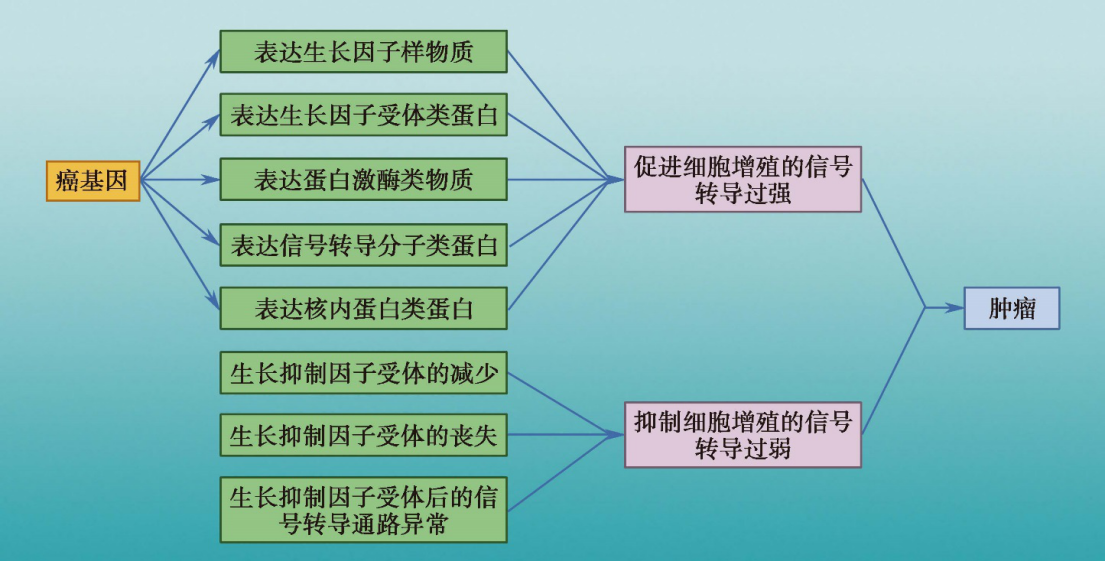
图10-10 恶性肿瘤细胞增殖过度的发生机制示意图
1. 促进细胞增殖的信号转导过强
(1)表达生长因子样物质 某些癌基因可以编码生长因子样的活性物质,例如,sis癌基因的表达产物与血小板源生长因子β链高度同源,int-2癌基因蛋白与成纤维细胞生长因子结构相似。此类癌基因激活可使生长因子样物质生成增多,以自分泌或旁分泌方式刺激细胞增殖。
(2)表达生长因子受体类蛋白 某些癌基因可以表达生长因子受体的类似物,通过模拟生长因子的功能受体起到促增殖的作用。例如,erb-B癌基因编码的变异型表达生长因子受体,缺乏与配体结合的膜外区,但可在没有生长因子存在的条件下持续激活下游的增殖信号。在人乳腺癌、肺癌、胰腺癌和卵巢肿瘤中已发现生长因子受体的过度表达;在卵巢肿瘤亦可见血小板源生长因子受体高表达,且这些受体的表达与预后呈负相关。
(3)表达蛋白激酶类 某些癌基因可通过编码非受体酪氨酸蛋白激酶或丝/苏氨酸激酶类影响细胞信号转导过程。例如,src癌基因产物具有较高的酪氨酸蛋白激酶活性,在某些肿瘤中其表达增加,可催化下游信号转导分子的酪氨酸磷酸化,促进细胞异常增殖。mos、raf癌基因编码丝/苏氨酸蛋白激酶类产物,其可促进MAPK磷酸化,进而促进核内癌基因表达。
(4)表达信号转导分子类 ras癌基因编码的小分子G蛋白Ras,可在Sos催化下通过与GTP结合而激活下游信号转导分子。在30%的人肿瘤组织已发现有不同性质的ras基因突变,变异的Ras与GDP解离速率增加或GTP酶活性降低,均可导致Ras持续活化,引起细胞生长失控而发生肿瘤。
(5)表达核内蛋白类 某些癌基因如myc、fos、jun的表达产物位于核内,能与DNA结合,具有直接调节转录活性的转录因子样作用。如高表达的Jun蛋白与Fos蛋白与DNA上的AP-1位点结合,激活基因转录,促进肿瘤发生。
2. 抑制细胞增殖的信号转导过弱
细胞癌变过程不仅可由促进细胞增殖的信号转导通路过强所致,还可能是生长抑制因子受体的减少、丧失以及受体后的信号转导通路异常,使细胞的生长负调控机制减弱或丧失。
转化生长因子β( transforming growth factor β, TGFβ)对多种肿瘤细胞具有抑制增殖及激活凋亡的作用,TGFβ受体(TβR)是具有丝/苏氨酸蛋白激酶活性的受体,分为I型和I型。I型受体与配体结合后,与I型受体形成寡聚体,并使I型受体磷酸化,激活的I型受体能使Smad蛋白家族的丝/苏氨酸残基磷酸化,之后Smad以二聚体的形式转人核内,调节靶基因的转录,通过抑制细胞周期素依赖性激酶4(CDK4)的表达,诱导P21wan、P27和P151nk4b等CDK抑制因子的产生,将细胞阻滞于G1期。
综上所述,细胞信号转导异常对疾病的发生发展具有多方面的影响。其发生原因也是多种多样的,如基因突变、细菌毒素、细胞因子、自身抗体和应激等均可造成细胞信号转导过程的原发性或继发性损伤。
细胞信号转导异常可以局限于单一环节,亦可同时或先后累及多个环节甚至多条信号转导途径,造成调节信号转导的网络失衡,引起复杂多变的表现形式。
细胞信号转导异常在疾病中的作用亦表现为多样性,既可作为疾病的直接原因,,引起特定疾病的发生;亦可干扰疾病的某个环节,导致特异性症状或体征的产生。细胞信号转导异常还可介导某些非特异性反应,出现在不同的疾病过程中。

