

第29课时 分子遗传标记的原理
一、第一代分子标记原理(理解)
1.RFLP分子标记:
(1)定义:以限制性片段长度多态性 (restriction fragment length polymorphism)作为分子标记,是发展最早的分子标记技术,该标记通过限制性内切酶酶切不同个体基因组DNA之后,采用Southern杂交技术获得含有与探针序列同源的酶切片段长度上的差异,进而开展多态性分析。
(2)原理:基因组DNA序列上的变化,如碱基替换、插入、缺失或重复,均会造成某种限制性内切酶(restrictionenzymes )酶切位点的增加或丧失以及内切酶酶切位点间DNA片段的插入、缺失或重复等,从而形成序列多态性。例如:
A、限制位点的碱基置换(Base substitution at the restriction sites):图A;
B、插入或删除(Insertions or deletions):图B。
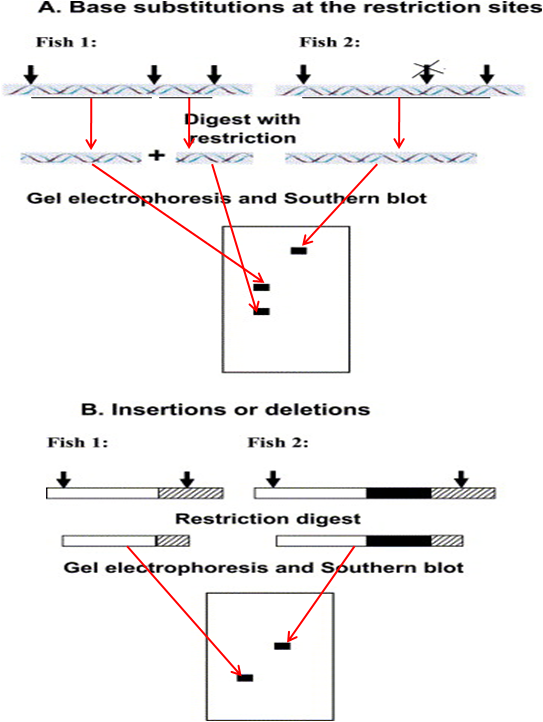
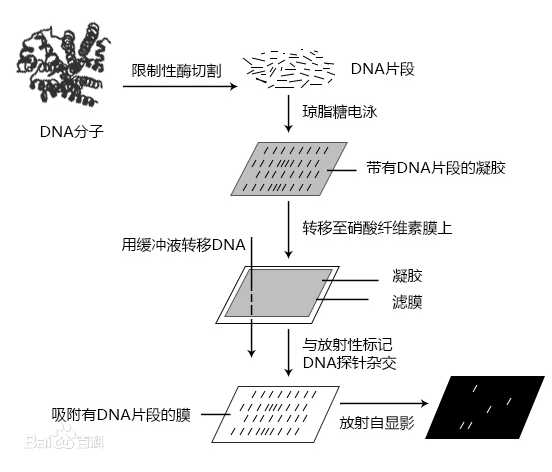
Southern杂交技术路线图
Southern印迹杂交是1975年由英国人southern创建的。Southern印迹杂交的基本方法是将DNA标本用限制性内切酶消化后,经琼脂糖凝胶电泳分离各酶解片段,然后经碱变性,Tris缓冲液中和和高盐下通过毛吸作用将DNA从凝胶中转印至硝酸纤维素膜上、烘干固定后即可用于杂交。附着在滤膜上的DNA与32P标记的探针杂交,利用放射显影术确立探针互补的每一条DNA带的位置,从而可以确定在众多消化产物中含某一特定序列的DNA片段的位置和大小。
(3)优点:
①无表型效应(不受发育阶段、器官和基因互作影响),且数量多;
②共显性,可以区分杂合子和纯合子(双亲的2个以上分子质量不同的多态性片段均在F1中表现);
③可以利用现有探针,具有种族特异性;
④遍及整个基因组;
⑤重复性好,稳定性高。
(4)缺点:
①DNA需要量大(5-30微克);
②步骤繁琐、周期长;
③检测效率低;
④同位素污染;
⑤需要制备多个探针;
⑥无法分析序列未明确的目标基因。
(5)技术路线图:
酶切→电泳→标记探针杂交→分析。
二、第二代分子标记原理(理解)
2.RAPD分子标记:
(1)定义:以随机扩增多态性DNA(Random AmplificationPolymorphism DNA,RAPD)为分子标记,该标记利用人工随机合成的寡聚脱氧核苷酸为引物(通常长度为10个核苷酸),PCR扩增基因组DNA,从而获得长度不同的多态性DNA片段,进而开展多态性分析。
(2)原理:RAPD是利用一系列不同碱基顺序的随机引物对所研究的基因组DNA进行PCR扩增,扩增产物(即DNA片段)通过琼脂糖或聚丙烯酰胺凝胶电泳分离。经特定的染料染色后在紫外检测仪上检测扩增产物的多态性,这些扩增的DNA片段长度的多态性反映了基因组相应区域的DNA序列多态性。
A图:引物结合部位的碱基置换(Base substitutions at the primer binding sites)引起的多态性;
B图:单碱基突变(single base mutation)引起的多态性。
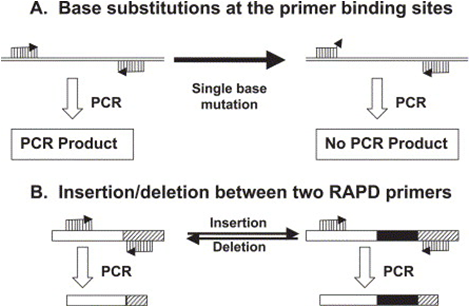

图.电泳图谱显示不同个体DNA序列多态性差异
(3)优点:
①丰富性大,能反映整个基因组的变化;
②极强的探测性,无需合成特定序列引物;高效性和灵敏性;
④技术简单,操作方便;
⑤所需样品DNA量少(10-100ng),质量要求低;
⑥环境污染小。
(4)缺点:
①稳定性低,同一反应不同次难重复,不同实验室难重复;
②显性遗传,无法区分纯合型与杂合型;
③高度变异性,即使在亲缘关系非常近的物种间,结果也造成很大差异。
(5)技术路线图:
提DNA→PCR→电泳→分析。
3.AFLP分子标记:
(1)定义:以扩增片段长度多态性(amplified fragmentlength polymorphism, AFLP)为分子标记,该标记选择性扩增基因组DNA酶切片段,获得限制性酶切片段的长度多态性,进而对多态性进行分析。
(2)原理:AFLP 是 RFLP 与RAPD相结合的产物,其基本原理是先利用限制性内切酶水解基因组 DNA 产生不同大小的DNA 片段,再使双链人工接头与酶切片段相连接,作为扩增(PCR)反应的模板DNA,然后以人工接头的互补链为引物进行预扩增,最后在接头互补链的基础上添加 1-3个选择性核苷酸作引物对模板 DNA 基因再进行选择性扩增,通过聚丙烯酰胺凝胶电泳分离检测获得的DNA扩增片段,根据扩增片段长度的不同检测出多态性。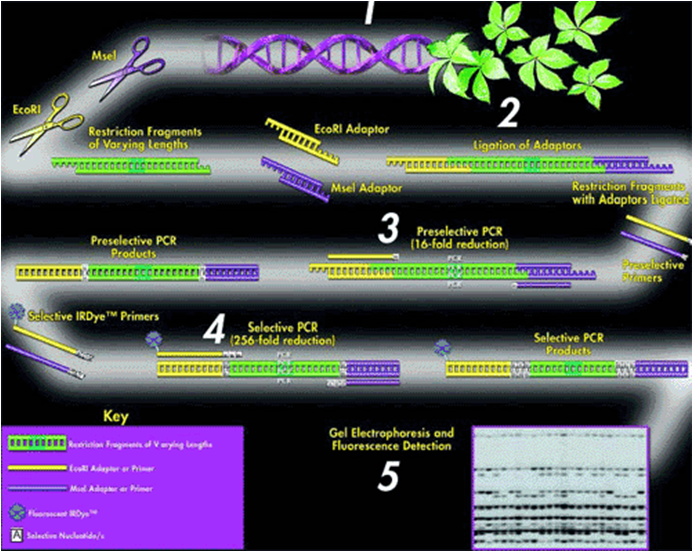
AFLP技术模式图(上、下)
.png")
 图.不同地理群体西施舌的AFLP电泳图谱
图.不同地理群体西施舌的AFLP电泳图谱
该技术的独特之处在于所用的专用引物在知道 DNA 信息的前提下就可对酶切片段进行PCR 扩增。为使酶切浓度大小分布均匀,一般采用两个限制性内切酶,一个酶为多切点,另一个酶切点数较少,因而 AFLP 分析产生的主要是由两个酶共同酶切的片段。
(3)优点:
①AFLP 结合了 RFLP 和 RAPD两种技术的优点,具有分辨率高、稳定性好、效率高的优点;
②标记数量多(内切酶和碱基组合可以调整);
③多态程度高(50-100带/次);
④典型的孟德尔方式遗传。
⑤无物种特异性;
⑥模板用量少;
⑦便于基因定位和克隆。
(4)缺点:
①成本高;
②内切酶质量要求高;
③DNA质量要求高。
尽管 AFLP 技术诞生时间较短,但可称之为分子标记技术的又一次重大突破,被认为是一种十分理想、有效的分子标记。这些技术在品种鉴别,目的基因定位及遗传图谱构建中得到了广泛的应用。
(5)技术路线图:
基因组DNA提取→ 限制性核酸酶双酶切基因组DNA→DNA片段两端连接上特定的接头→ 选择扩增→ 聚丙烯酰胺凝胶电泳→ 凝胶转移,干胶处理→自显影。
4.SSR分子标记:
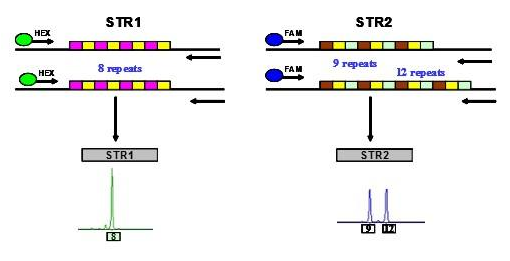
(1)定义:以简单重复序列(Simple Sequence Repeats, SSRs),又称微卫星DNA(microsatellite DNA)或短串联重复序列(Short Tandem Repeat ,STR)为分子标记,该标记根据微卫星DNA两端的单拷贝序列设计一对特异引物,利用PCR技术,扩增每个位点的微卫星DNA序列,通过电泳获得核心序列的长度多态性,并对其多态性进行分析。
(2)原理:微卫星DNA重复单位序列在卫星DNA分类中属于最短,由几个(1-10个)核苷酸组成重复单位,只有2~6bp,重复单位串联成簇,长度50~100bp。如(CA)n、(TG)n、(AT)n、(CC)n、(GATA)n;n代表重复次数。每个SSR两侧的序列一般是相对保守的单拷贝序列(单一序列)。单拷贝序列在基因组中占50-80%,如人基因组中,大约有60-65%的序列属于这一类(下图)。

拷贝序列中储存了巨大的遗传信息,编码各种不同功能的蛋白质。不同生物基因组中单拷贝序列和重复序列的比例差异很大。真核生物基因组中有许多未知功能的重复序列。SSR均可被转录,有些编码蛋白质,而另一些则位于非转译区的5′端和3′端不编码蛋白质。
SSR标记根据微卫星DNA两端的单拷贝序列设计一对特异引物,利用PCR技术,扩增每个位点的微卫星DNA序列,通过电泳获得核心序列的长度多态性。
(3)技术路线图:微卫星序列的获取(NCBI、测序等)→设计引物→DNA提取→PCR扩增→聚丙烯酰胺凝胶电泳(下图)→分析。
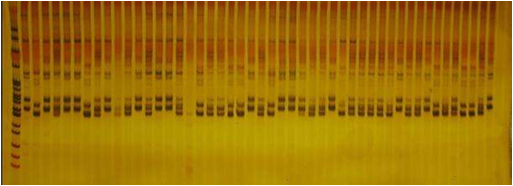
(4)优点:
①SSR数量多,分布整个基因组,多态性高;
②检测到的为单一多等位基因位点;
③共显性,可鉴定出杂合子和纯合子;
.png")

④重复性好,稳定性高;
⑤DNA样品量少,且质量要求不高。
(5)缺点:
①需要预知DNA序列;
②引物设计工作量大。
三、第三代分子标记(理解)
5.SNP分子标记:
(1)定义:以单个核苷酸的变异,包括单碱基的转换、颠换、插入、缺失等所引起的DNA序列多态性,即单核苷酸多态性(single nucleotide polymorphism, SNP) 为分子标记,该标记利用数据库中已有的SNP对特定种群的序列和发生频率开展研究,采用基因芯片技术、Taqman技术、分子信标技术和焦磷酸测序法,进行基因分型,从而获得单核苷酸多态性信息,并对其多态性进行分析。
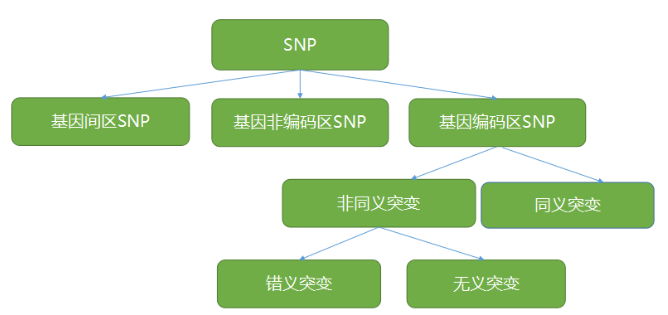
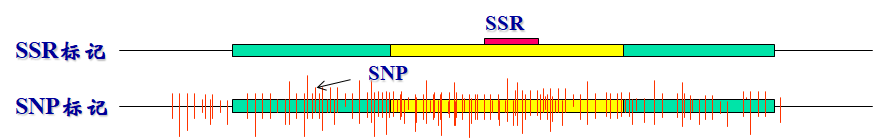
图.两种标记的区别
(2)原理:是指利用数据库中已有的SNP进行特定种群的序列和发生频率的研究,主要包括基因芯片技术,Taqman技术,分子信标技术和焦磷酸测序法等,从而获得基因分型。
①基因芯片技术:是在固相支持介质上进行分子杂交和原位荧光检测的一种高通量SNP分析方法。优点是高通量,一次可对多个SNP进行规模性筛选,被捡起材料也很少,操作步骤简单。缺点是芯片设计成本高,由于DNA样品的复杂性,有些SNP不能被捡起。
②Taqman技术:在pcr反应系统中加入2种不同荧光标记的探针,它们分别与两个等位基因完全配对。探针运用了荧光共振能量转换技术,探针的5’端和3’端分别用特殊染料标记,称为供体-受体染料对或爆-猝灭燃料对。
③分子信标技术:与Taqman技术相似,分子信标技术也是在PCR反应体系中加入荧光标记的探针与靶序列杂交,通过仪器检测荧光值的变化,进行基因分型。优点是分子信标法在选择荧光染料时,不必像Taqman法那样考虑染料之间光谱的重叠性,一次可以使用4种或4种以上染料,同时对多个SNP进行分析;Taqman法和分子信标法优点都是简单操作,可以自动化。缺点是不能达到高通量分析,荧光探针费用高。
④焦磷酸测序法:由4种酶催化同一反应体系中的酶级联反应,包括DNA聚合酶、硫酸化酶、荧光素酶和双磷酸酶,反应底物为adenosine5’phosphosulfate 和荧光素,是一种不依赖平板胶或毛细管电泳,不依赖DNA的荧光标记/激发/检测体系的序列分析技术,适用于已知SNP的序列验证及基因分型。
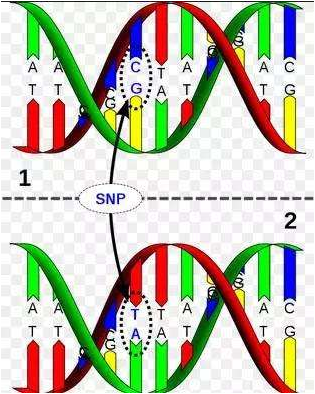
理论上讲,SNP既可能是二等位多态性,也可能是3个或4个等位多态性,但实际上,后两者非常少见,几乎可以忽略。因此,通常所说的SNP都是二等位多态性的。这种变异可能是转换,也可能是颠换。转换的发生率总是明显高于其它几种变异,具有转换型变异的SNP约占2/3,其它几种变异的发生几率相似。
下述为SNP变异来源-碱基置换:
①转换transition:AT→GC及GC→AT
一种嘧啶置换另一种嘧啶 T↔C
一种嘌呤置换另一种嘌呤 A↔G 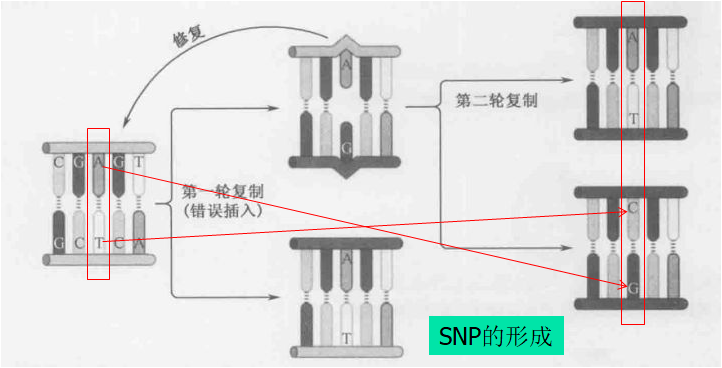
没有被及时除去的突变可以通过下一轮复制永久插入基因组,形成SNP。
②颠换 transversion:AT→TA或CG,GC→CG或TA 嘌呤与嘧啶互换,C↔A,A↔T等

③缺失/插入:不常见
碱基置换的后果可能是:
①同义突变(silent mutation),位于密码子第三碱基的置换,由于遗传密码的简并,经转录和翻译所对应的氨基酸不变。
②错义突变(missense mutation),碱基置换使密码子的意义改变,经转录和翻译所对应的氨基酸改变。
③无义突变(nonsense mutation),碱基置换使密码子成为终止密码,导致肽链延长提前结束。
④终止密码突变(terminator codon mutation),碱基置换使终止密码转变成某种氨基酸密码,指导合成的肽链将延长到出现第二个终止密码才结束。
(3)技术路线图:
提取基因组DNA→设计并合成SNP引物→PCR扩增→分型检测→直接测序→与已知序列进行比对→液相质谱→高分辨率溶解曲线。
(4)优点:
①密度高-SNP在人类基因组的平均密度估计为 1\1000bp , 可以在任何一个待研究基因的内部或附近提供一系列标记。
②富有代表性-某些位于基因内部的SNP 有可能直接影响蛋白质结构或表达水平,因此,它们可能代表疾病遗传机理中的某些作用因素。
③遗传稳定-与微卫星等重复序列多态性标记相比,SNP具有更高的遗传稳定性。
④易实现分析的自动化-SNP标记在人群中只有两种等位型,所以它是一种二态的标记,即二等位基因(biallelic)。 由于SNP的二态性,非此即彼,检测时只需一个“ + \- ”或“全\无”的方式,检测分析方法易实现自动化。
(5)缺点:
对测序的精度要求高。
6.DNA条形码:
(1)定义:DNA条形码(DNA barcode)是指生物体内能够代表该物种的、标准的、有足够变异的、易扩增且相对较短的DNA片段,是一个物种的标记。该标记以传统的DNA测序技术为基础,通过对一组来自不同生物个体的短的同源DNA序列(约800bp)进行PCR扩增和测序,对测得的序列进行多重序列比对和聚类分析,从而将某个个体精确定位到一个已描述过的分类群中,实现DNA序列和生物物种之间的一一对应关系。
DNA条形码技术是利用生物体DNA中一段保守片段对物种进行快速准确鉴定的新兴技术。2003年,全球多位生物学专家深入讨论了DNA条形码的科学性和社会功能,提出了国际DNA条形码计划。
DNA条形码已经成为生态学研究的重要工具,不仅用于物种鉴定,同时也帮助生物学家进一步了解生态系统内发生的相互作用。在发现一种未知物种或者物种的一部分时,研究人员便描绘其组织的DNA条形码,而后与国际数据库内的其他条形码进行比对。如果与其中一个相匹配,研究人员便可确认这种物种的身份。

(2)原理:在DNA分类学(DNA Taxonomy,即以DNA序列作为生物分类系统平台)的基础上,针对形态学分类固有的缺陷,如表型可塑性和遗传可变性、无法鉴定隐存分类单元和不同发育阶段的物种等,加拿大动物学家Paul Hebert等对动物界,包括脊椎动物和无脊椎动物共11门13320个物种的线粒体细胞色素C氧化酶亚基基因序列进行比较分析,除腔肠动物Cnidaria外,98%的物种遗传距离差异在种内0%~2%,种间平均可达到11.3%,据此提出可以用单一的小片段基因来代表物种,作为物种的条形编码,为全球生物编码,即DNA条形码(DNAbarcoding)。它是利用一段标准DNA 序列作为标记来实现快速、准确和自动化的物种鉴定,类似于超市利用条形码扫描区分成千上万种不同的商品。因此,Hebert等在2003年提出了DNA条形码的概念,首先倡导将条形码编码技术应用到生物物种鉴定中,因此他被称为DNA条形编码之父。
理想的DNA barcoding应当符合下列标准:
①具有足够的变异性以区分不同的物种,同时具有相对的保守性;
②必须是一段标准的DNA区来尽可能鉴别不同的分类群;
③目标DNA区应当包含足够的系统进化信息以定位物种在分类系统(科、属等)中的位置;
④应该是高度保守的引物设计区以便于通用引物的设计;
⑤目标DNA区应该足够的短以便于有部分降解的DNA的扩增。
NA barcoding作为生物“种水平species-level”鉴定的工具引人注目。Genbank数据库中CO I序列(细胞色素c氧化酶亚基 I)正在快速增加。Min等分析了CO I序列及其来源基因组核苷酸含量之间的关系,结果表明849个CO I基因的5端的DNA barcoding序列令人惊奇地准确地代表了其来源完整线粒体基因mtDNA的重要信息,也就是说对于未测序的基因组,从DNA barcoding能快速预知完整基因组的组成 。
(3)技术路线:
提取基因组DNA→合成引物→PCR扩增→克隆→测序分析
(4)优点:
①准确性高,可以鉴别形态上高度相似或表型性状可塑性高的物种;
②通过建立DNA条形码数据库,汇聚全球的物种资料,加速新种和隐存种的发现;
③简单、快速、通量高的鉴定物种。
(5)缺点:
DNA条形码提出之初,强调用统一的基因(线粒体CO I)序列进行鉴定物种,如果进化过慢,则难以鉴定。目前多基因分析法被普遍接受,必要时还要和传统的分类方法相结合。
(6)DNA条形码分子鉴定常用方法:
①ITS2:
ITS(internal transcribed spacer of nuclear ribosomal DNA),细胞核的核糖体DNA内部转录间隔区,是码核糖体RNA前体基因的一段,位于核仁区,多用来进行进化分析。ITS位于18S rRNA基因和28S rRNA基因之间,中部被5.8S rRNA基因一分为二,即ITS1(the first internal transcribed spacer)区和ITS2(the second internal transcribed spacer)区。5.8S、18S和28S进化速率较慢,常用于探讨科级和科级以上等级的系统发育问题。而间隔区ITS(包括ITS1和ITS2)进化速率较快,一般用于研究属间、种间甚至居群间等较低分类等级的系统关系。
②psbA-trnH:
psbA-trnH基因间区是位于叶绿体基因psbA基因和trnH基因之间的一段非编码区,该间区进化速率较快,常用于植物属间、种间的系统发育研究。
③CO I:
CO I为线粒体基因组的蛋白质编码基因,全称为细胞色素c氧化酶亚基 I (cytochrome c oxidase subunit I),由于该基因进化速率较快,常用于分析亲缘关系密切的种、亚种及地理种群之间的系统关系。
四、几种分子标记的综合比较(理解)

复习题:
一、问答题:
1.试述AFLP分子标记的原理及其优缺点。
2.试述SNP分子标记的原理及其优缺点。
3.试述DNA条形码分子鉴定常用的方法。
二、简答题:
1.试述RFLP分子标记的原理及其优缺点。
2.试述RAPD分子标记的原理及其优缺点。
3.试述SSR分子标记的原理及其优缺点。
4.试述SNP变异的主要来源。
5.试述DNA条形码的原理及其优缺点。
6.请对几种分子标记进行综合比较。
三、名词解释:
1.RFLP分子标记;
2.Southern印迹杂交;
3.RAPD分子标记;
4.AFLP分子标记
5.SSR分子标记;
6.SNP分子标记;
7.基因芯片技术;
8.DNA条形码。

