免疫缺陷病(immunodeficiencydisease.IDD)是因遗传因素或其他原因造成免疫系统先天发育障碍或后天损伤所致的综合征。患者因免疫细胞发育、分化、增生、调节和代谢异常。出现一系列临床表现:对病原体(细菌、病毒、真菌)甚至条件性病原微生物高度易感:对自身免疫病及超敏反应性疾病易感:某些肿瘤特别是淋巴细胞恶性肿瘤的发生率增高。免疫缺陷病按病因不同分为原发性免疫缺陷病(primary immuno deficiency disease, PIDD)和获得性免疫缺陷病(acquired immuno deficiency disease AIDD)两大类。
第一节 原发性免疫缺陷病
PIDD 又称为先天性免疫缺陷病(congenital immuno deficiency disease.CIDD).由免疫系统遗传缺陷或先天发育不全所致,多于幼年起病。2011年WHO 和国际免疫学联合会(IUIS)联合组织会议将PIDD分为八大类,即T、B细胞联合免疫缺陷病、以抗体缺陷为主的免疫缺陷病,吞噬细胞数量和(或)功能先天性免疫缺陷病、补体缺陷病、已经定义明确的免疫缺陷病、免疫失调性免疫缺陷病、固有免疫缺陷病和自身炎性反应性疾病引起的免疫缺陷病。PIDD已经超过350种,每年有超20种新发现疾病。
一、T、B 细胞联合免疫缺陷病
联合免疫缺陷病(combined immuno deficiency disease CID)是同时累及机体细胞免疫和体液免疫的PIDD。T、B细胞分化发育中涉及多种分子,其中任一分子的基因突变都可引起免疫缺陷病。重症联合免疫缺陷病(severe combined immuno deficiency disease,SCID)由T细胞发育异常和(或)B细胞发育不成熟引起,包括 T-B-SCID、T-B+SCID等20多种疾病。SCID多见于新生儿和婴幼儿,易发生肺炎、脑膜炎等严重感染。但某些SCID患者表现为慢性皮疹,是由于母亲T细胞进入胎儿而未被排斥(胎儿缺乏T/B细胞或其功能)导致移植物抗宿主反应,即母亲T细胞对胎儿组织发生免疫攻击。能引起SCID的主要突变基因有 IL-2RG、JAK3、IL-7Rα、RAGI、RAG2、DCLRE1C、Ligase4、DNA-PKcs、CD3δ、CD3ε、CD3εξ、ADA和CD45,各个基因突变引起的疾病表型不同。
(一)T细胞缺陷、B细胞正常的重症联合免疫缺陷病(T-B+SCID)
T-B+SCID患者的血液中T细胞显著减少,NK细胞减少或正常,B细胞数量正常但血清lg降低。X性联重症联合免疫缺陷病占T-B+SCID的40%,是细胞因子受体亚单位γc链缺陷所致,为X连锁遗传。临床表现为出生后不久即发生严重呼吸道感染、慢性腹泻和夭折。γc链是IL-2、IL-4、IL-7等细胞因子受体共有亚单位,介导多种细胞因子的信号转导从而调控T、NK细胞分化和成熟,其基因突变使T细胞和NK细胞发育停滞。因此尽管B细胞数量正常,但由于缺乏T细胞的辅助,体液免疫功能仍然缺陷。
(二)T、B细胞均缺如的重症联合免疫缺陷病(T-B-SCID)
T-B-SCID为常染色体隐性遗传,特征为循环淋巴细胞极度减少、各种Ig缺乏。腺苷脱氨酶(adenosinedeaminase,ADA)缺陷占SCID的10%~15%,ADA缺陷使细胞内dATP或dGTP积聚,抑制 DNA合成所必需的核糖核苷还原酶,影响淋巴细胞生长和发育,临床表现为免疫系统缺陷病的典型特征,还可见耳聋、行为障碍、肋软骨异常和肝毒性等症状。导致T-B-SCID的缺陷基因还包括重组活化基因(RAG1/RAG2)等。
二、以抗体缺陷为主的原发性免疫缺陷病
这是一类以抗体生成及抗体功能缺陷为特征的疾病,患者一般有血清Ig减少或缺乏,出生后7~9月龄开始发病,患儿对肿瘤和自身免疫病易感,对有荚膜的化脓性细菌易感,但对真菌和病毒则不易感。这类疾病包括:①血清Ig和B细胞显著降低或缺失型;②至少两类血清Ig显著降低伴B细胞功能正常或降低型:③血清IgG、IgA显著降低伴IgM正常或升高伴B细胞数目正常型;④Ig同种型缺陷或轻链缺陷伴B细胞数目正常型:⑤特异性抗体缺陷伴Ig水平正常和B细胞数目正常型;⑥婴儿暂时性低丙种球蛋白血症。发病机制为:参与B细胞分化发育过程的信号分子基因,包括 Btk、TACI、λ5、Igα、Igβ、BLNK、ICOS、CD19、CD81、CD20、CD40、K等缺陷,导致B细胞停留在分化发育某一阶段,成熟B细胞数量减少或功能缺陷,引起抗体生成及功能缺陷。
(一)X连锁无丙种球蛋白血症(X-linked agammaglobulinemia,XLA)
X连锁无丙种球蛋白血症即Bruton病.特点是:外周成熟B细胞、浆细胞及各类Ig显著降低或缺,但原始B细胞和细胞数量及功能正常。多见于出生6~9个月男性婴出现反复化脓性细药感染;注射丙种球蛋白能控制感染,但因无法诱导呼吸道 SIgA使鼻部、肺部感染极易复发;发病机制是Btk基因突变。Btk分子参与未成熟B细胞分化和成熟B细胞活化。Btk基因突变或缺失致酪氨酸激酶合成障碍.B细胞发育停滞于前B细胞状态,导致成熟B细胞数目减少甚至缺失。
(二)普通变异型免疫缺陷病(common variable immunodeficiency,CVID)
CVID是一种常见的低丙种球蛋白血症,又称成人型或迟发性低丙种球蛋白血症,为一组遗传方式不定、病因不明确、主要影响抗体合成的PIDD。大多数 CVID是由于T细胞功能异常不能提供有效辅助,导致B细胞不能合成抗体和发生类别转换。患者体内lgG和lgA水平明显降低,IgM 可能正常或下降,伴B细胞数量正常或降低,但较XLA为轻。临床表现多样,常发病于学龄期和成人期,易患反复细菌感染,部分有自身免疫病、淋巴组织增生和(或)肉芽肿病。
三、吞噬细胞数量和(或)功能先天性免疫缺陷病
这类疾病包括中性粒细胞分化缺陷、运动缺陷、呼吸爆发缺陷、对分枝杆菌病的遗传易感缺陷及其他缺陷五种疾病。临床表现为化脓性细菌和真菌的反复感染,轻者仅累及皮肤,重者则感染重要器官而危及生命。新发现的相关缺陷基因有:p40、phox、gp91、phox、IRF8、TAZ、COHI、C16/f57、GATA2。
(一)X连锁慢性肉芽肿病(X-linked chronic qranulomatous disease,CGD)
CGD是常见的吞噬细胞功能缺陷性疾病,因呼吸爆发缺陷所致。患者多数为X连锁遗传,多为男性,表现为反复、严重的化脓性感染,在淋巴结、肺等多器官形成化脓性肉芽肿,并伴有反应性高丙种球蛋白血症。CGD病因是细胞色素b-B亚单位(CYBB)基因突变,导致中性粒细胞、单核巨噬细胞缺乏NADPH氧化酶,不能有效杀灭被吞噬菌,后者持续存活并随吞噬细胞游走播散至全身。慢性感染可引起吞噬细胞在局部聚集,并持续刺激CD4+T细胞以招募和激活更多巨噬细胞,从而形成肉芽肿。IFN-γ被用于 CGD 的临床治疗。
(二)孟德尔式易感分枝杆菌病(Mendelian susceptibility to mycobacterial disease, MSMD)
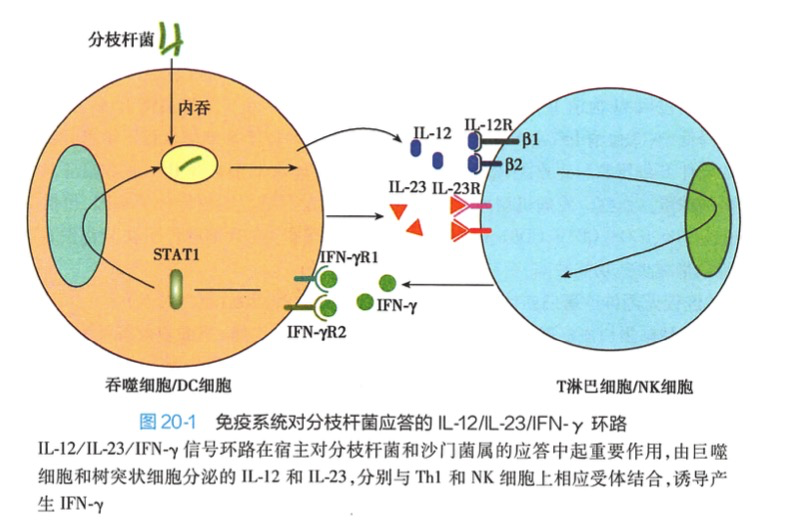
MSMD是一种由IL-12/IL-23/IFN-γ及其受体,或信号转导分子缺陷引起的罕见常染色体隐性遗传性综合征,MSMD患者易受弱毒力分枝杆菌属如卡介苗、非结核分枝杆菌、环境分枝杆菌等感染,对结核分枝杆菌更易感。分枝杆菌为胞内菌,宿主抗胞内菌感染主要依赖细胞免疫应答。DC和巨噬细胞经由TLRs识别分枝杆菌的PAMP 而被活化,产生IL-12和IL-23等细胞因子,激活Th、NK分泌IFN-γ、IL-17和TNF-a等细胞因子;IFN-γ进一步增强巨噬细胞的抗原提呈和杀伤病原体能力,如此形成IL-12/IL-23/IFN-γ环路。MSMD是此环路参与基因如IL-12p40、IL-12RβI、IFN-γ受体、STAT1等缺陷,导致巨噬细胞和T细胞对胞内菌的杀伤作用减弱甚至消失,因而易发生分枝杆菌等胞
内菌感染。
四、补体缺陷病
补体缺陷病多为常染色体隐性遗传,由补体固有成分、调节蛋白或补体受体中任一成分缺陷引起。补体固有成分缺陷患者表现为 SLE 样综合征、抗感染能力低下、易发生化脓性细菌(如奈瑟菌)感染。补体调节蛋白或补体受体缺陷者表现为抗感染能力降低。
(一)遗传性血管神经性水肿
遗传性血管神经性水肿为常见补体缺陷病,由CIINH基因缺陷所致。这种补体调节蛋白缺乏引起C2裂解失控,C2a产生过多,导致血管通透性增高。患者表现为反复发作的皮肤黏膜水肿,若水肿发生于喉头可导致窒息死亡。
(二)阵发性夜间血红蛋白尿
阵发性夜间血红蛋白尿的发病机制是编码糖基磷脂酰肌醇(glycosyl phosphatidvlinositol, GPI)的pig-a基因翻译后修饰缺陷。补体调节成分衰变加速因子(DAF/CD55)和膜反应性溶破抑制物(MIRL/CD59)是补体溶细胞效应的抑制因子,它们通过GPI锚定在细胞膜上。由于GPI合成障碍,患者红细胞不能锚定DAF和MIRL而发生补体介导的溶血。临床表现为慢性溶血性贫血、全血细胞减少和静脉血栓形成,晨尿出现血红蛋白。
五、已经定义明确的免疫缺陷病
这类疾病包括Wiskott-Aldrich综合征(WAS)、DNA修复缺陷病等9种疾病。除WAS和角化不良素基因(DKCI)突变引起的Hoyeraal-Hreidarsson综合征为X连锁遗传外,其余均为常染色体遗传。可引起该类疾病的突变基因有WASP、ATM、NBSI、PMS2、RMRP、STAT3、SPII0、DKCI、IKAROS等。
Wiskott-Aldrich综合征(WAS)
WAS即伴湿疹和血小板减少的免疫缺陷病.为X连锁遗传.发病机制是X染色体编码的 WAS蛋白(Wiskott-Aldrich syndrome protein,WASP)基因缺陷。WASP表达于造血细胞,能调节细胞骨架的重排.使T细胞和APC间形成免疫突触以活化T细胞。WASP基因缺陷使细胞骨架不能移动则免疫细胞间相互作用受阻,导致T细胞活化及功能障碍。WAS多见于男性婴儿,表现为反复细菌感染、血小板减少症和皮肤湿疹,可伴发自身免疫病和恶性肿瘤。同时存在血小板减少、反复感染、湿疹三联症者占27%。
六、免疫失调性免疫缺陷病
免疫失调性免疫缺陷病包括免疫缺陷伴色素减退、家族性嗜血淋巴组织细胞增多(familial he- mophagocyticlymphohistiocytosis,FHL)综合征、X连锁淋巴组织增生综合征(X-linked lymphoproliferative.XLP)及自身免疫综合征四种疾病。除SH2DIA、XIAP基因突变引起的 XLP和Foxp3 基因突变引起的自身免疫综合征为X连锁遗传外,其余均为常染色体遗传。
免疫系统通过不同机制使经抗原活化的已经发生偏移的克隆库或 TCR/BCR 受体库回复到稳定状态。即抗原被清除后,机体通过 Fas/FasL途径、TNFα信号转导途径诱导AICD而控制活化淋巴细胞数量,发挥免疫自稳作用。因此上述途径中相关基因缺陷都会引起免疫失调性疾病。目前发现,编码 Fas分子及其下游途径的 TNFRSF6、TNFSF6、CASP10、CASP8、NRAS 基因突变可引起 XLP。
七、固有免疫缺陷病
固有免疫缺陷病包括无汗性外胚层发育不良伴免疫缺陷(ectodermal dysplasia with immunodefi- ciencyEDA-ID)、IL-1受体相关激酶4(IL-1 receptor associated kinase4,IRAK4)缺陷等多种疾病。
无汗性外胚层发育不良伴免疫缺陷
EDA-ID是一类因NF-KB活化关键调节因子(NF-KBessential modulator,NEMO)基因突变导致的发育缺陷病综合征。患者多为男性,表现为少汗或无汗、对热的耐受性差、毛发稀疏、无牙或少牙、反复发生化脓性细菌威染。NF-B在静自状态下以无活性形式存在.上游信号刺激诱导 NF-KB抑制蛋白(IkB)发生磷酸化.促进NF-KB蛋白二聚体形成并进入胞核,激活并参与基因转录。NEMO是调节NF-KB功能的关键因子,当发生错义突变后,IKB不能发生磷酸化,NF-KB及其相关信号通路活化受阻,进而引起经典型 EDA-ID。
八、自身炎性反应性疾病引起的免疫缺陷病
自身炎性反应性疾病引起的免疫缺陷病包括涉及和未涉及炎症小体的两种免疫缺陷疾病。其发病机制是参与NF-KB 信号途径、细胞凋亡及IL-1β分泌过程中的信号分子基因如MEFV、MVK、CIASI、NLRP12、TNFRSFIA、L-10/IL-10R、PSTPIP1/C2BP1、NOD2、LPIN2、LPINI等突变引起信号转导紊乱。

