
苯丙酮尿症
苯丙酮尿症(phenylketonuria,PKU)是一种常染色体隐性遗传病,是由于苯丙氨酸羟化酶(phenylalanine hydroxylase,PAH)缺乏或其辅酶四氢生物蝶呤(tetrahydrobiopterin,BH4)缺乏,导致血苯丙氨酸(phenylalanine,Phe)增高的一组最常见的氨基酸代谢病,也称高苯丙氨酸血症(hyperphenylalaninemia,HPA)。PKU是先天性氨基酸代谢障碍中最为常见的一种,临床表现为智力发育落后,皮肤、毛发色素浅淡和鼠尿样体味。其发病率有地区和种族差异,我国发病率为1:11000,北方人群高于南方人群。
【病因及发病机制】
苯丙氨酸是人体必需氨基酸,摄入体内的Phe一部分用于蛋白质的合成,部分通过苯丙氨酸羟化酶作用转变为酪氨酸,仅有少量的Phe经过次要代谢途径,在转氨酶的作用下转变成苯丙酮酸,其代谢途径见图17-3。
图17-3 苯丙氨酸主要代谢途径
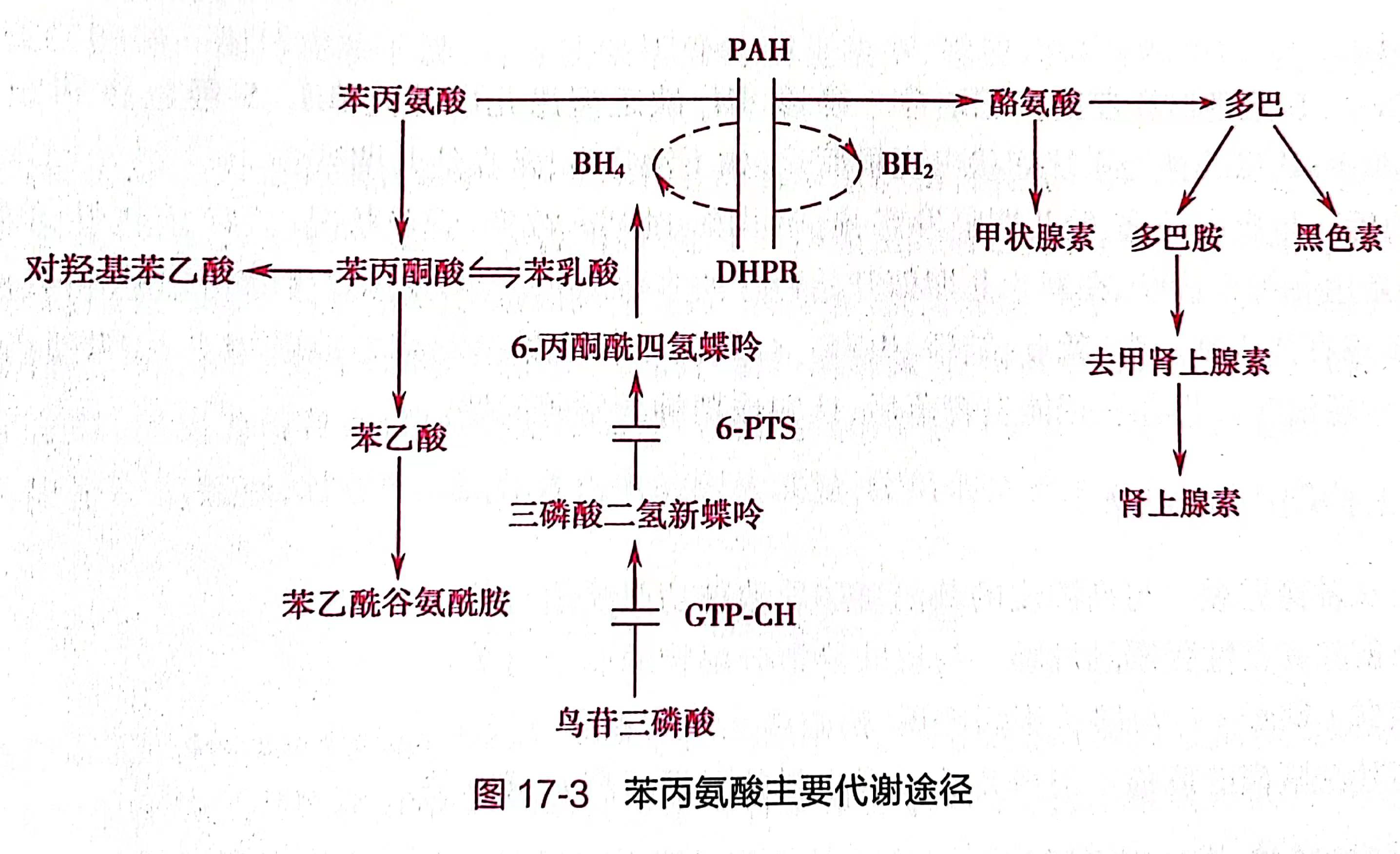
本病按酶缺陷不同可大致分为典型PKU和BH4缺乏型PKU。
1.典型PKU 绝大多数患儿为典型病例。由于患儿肝细胞缺乏PAH,不能将Phe转化为酪氨酸,导致Phe在血液、脑脊液及各种组织液和尿液中的浓度极高,同时通过旁路代谢产生大量苯丙酮酸、苯乙酸、苯乳酸和对羟基苯乙酸。高浓度的Phe及其旁路代谢产物可导致脑损伤。同时,由于酪氨酸生成减少,致使甲状腺素、肾上腺素和黑色素等合成不足。
2.BH4缺乏型PKU 是由鸟苷三磷酸环化水合酶、6-丙酮酰四氢喋呤合成酶或二氢生物喋呤还原酶等酶缺乏所致。BH4是苯丙氨酸、酪氨酸和色氨酸等芳香氨基酸在羟化过程中所必需的共同的辅酶,BH4缺乏时不仅苯丙氨酸不能氧化成酪氨酸,而且造成多巴胺、5-羟色氨等重要神经递质的合成受阻,加重了神经系统的功能损害,故BH4缺乏型PKU的临床症状更重,治疗亦不易。我国新生儿筛查中发现的高苯丙氨酸血症患儿中,约10%~15%为BH4缺乏症。
【临床表现】
患儿出生时都正常,一般在3~6个月时开始出现症状,后逐渐加重,1岁时症状明显。
1.神经系统表现 以智能发育落后为主,可有表情呆滞、行为异常、多动、肌痉挛或癫痫小发作。少数呈肌张力增高和腿反射亢进,80%有脑电图异常。BH4缺乏型PKU患儿的神经系统症状出现较早且较重,肌张力明显减低,嗜睡或惊厥,智能明显落后。
2.外貌 生后数月因黑色素合成不足,毛发由黑变黄,皮肤和虹膜色泽变浅。皮肤干燥,常有湿疹。
3.体味 由于尿及汗液中排出较多苯乙酸,有明显的鼠尿样臭味。
4.其他 可有呕吐、喂养困难。PKU母亲在未控制血苯丙氨酸浓度的情况下怀孕,其子女即使不是PKU,也常伴有小头畸形和智力低下。
【辅助检查】
1.新生儿筛查 新生儿哺乳2~3日后,采集足跟血液,滴于专用采血滤纸上,晾干后即可寄送至筛查实验室,进行Phe测定。当Phe大于切割值时,应进一步检查和确诊。
2.苯丙氨酸浓度测定 正常浓度<120umol/L(2mg/dl),典型PKU>1200umol/L,中度PKU为360~1200umol/L,轻度HPA为120~360umol/L。
3.尿蝶呤图谱分析和DHPR活性测定 主要用于BH4缺乏症的鉴别诊断。
4.DNA分析 用DNA分析方法作基因突变检测,进行基因诊断和产前诊断。
【治疗要点】
疾病一旦确诊,应立即治疗,开始治疗的年龄愈小,效果愈好。开始治疗的理想时间是出生后1周内。
1.低苯丙氨酸饮食 为主要治疗手段,每日Phe按30~50mg/kg供给,维持血Phe浓度在120~360umol/L为宜,使摄入Phe的量能保证生长发育和体内代谢的最低需要。饮食治疗对象包括典型PKU和血Phe浓度持续超过360umol/L者。对于血Phe浓度在120~360umol/L者,目前不主张饮食限制。
血Phe理想控制浓度范围为:1岁以内,120~240umol/L;1~12岁,120~360umol/L;>12岁,120~600umol/L。
2.BH4、5-羟色氨酸和左旋多巴治疗 确诊BH4缺乏型患儿应补充BH4、5-羟色胺和左旋多巴。BH4缺乏型PKU患儿神经系统症状出现早而重,如不治疗者,常在幼儿期死亡。
3.预后 与病情轻重、胎儿期脑发育、治疗早晚、血Phe浓度、营养状况、治疗依从性等多种因素有关。经新生儿筛查诊断,在新生儿期即开始治疗的多数患儿,智力及体格发育可以达到或接近正常水平,很多患者能正常上学、就业、结婚、生育。合理的个性化饮食治疗是改善患儿远期预后的关键,但少数患者经治疗后仍存在智能发育落后、认知和精神异常的问题。
【常见护理诊断/问题】
1.生长发育迟缓 与高浓度的苯丙氨酸导致脑细胞受损有关。
2.有皮肤完整性受损的危险 与皮肤异常分泌物的刺激有关
3.焦虑(家长) 与缺乏疾病知识、担心患儿疾病预后有关。
4.有社交隔离的危险 与患儿的外观引起的恐惧或窘迫感有关。
【护理措施】
1.饮食护理 PKU患者PAH酶活性不同,导致对Phe耐受量的个体差异,应根据不同年龄段患儿每日蛋白质需要量、血Phe浓度、Phe的耐受量、饮食嗜好等个体化调整饮食治疗方法。
(1)新生儿和婴儿期:喂养应以乳类为主。经典型PKU患儿可暂停母乳或普通婴儿奶粉,给予无Phe特殊奶粉,治疗3~7天后血Phe浓度下降接近正常后,逐步添加少量天然乳品,首选母乳(母乳中Phe含量仅为牛奶的1/3)。轻度PKU根据血Phe浓度将无Phe特殊奶粉与普通奶粉按3 : 1或2 : 1混合配制,并根据血Phe浓度调节饮食搭配。
(2)幼儿及儿童期:为满足蛋白质需要及血Phe浓度控制,可选则无Phe蛋白粉和/或奶粉,并根据个体Phe耐受量,参考常用食物的Phe含量选择不同的天然食物(表17-1)。日常饮食中应避免Phe含量较高的食物(如肉、乳酪、鱼、蛋、面粉、坚果、豆制品),可适当食用Phe含量中等的食物(包括大米、牛奶、早餐麦、土豆、奶油)或Phe含量较低的淀粉类食物、水果、蔬菜等。
(3)青少年及成年期:鼓励该期患者坚持治疗,有效控制血Phe浓度。对PKU女性患者需进行产前遗传咨询,在孕前6个月至整个孕期需要饮食治疗,控制在120~360umol/L以下直至分娩,以免孕期血Phe浓度增高而导致胎儿脑发育障碍及各种畸形发生。
治疗时应定期监测血中苯丙氨酸浓度,1岁以内每周1次,1~12岁每个月1~2次,12岁后每1~3个月1次,孕期每周2次,同时注意生长发育情况。
表17-1 常用食物的苯丙氨酸含量(每100g食物)
| 食物 | 蛋白质(g) | 苯丙氨酸(mg) | 食物 | 蛋白质(g) | 苯丙氨酸(mg) |
| 人奶 | 1.3 | 36 | 藕粉或麦淀粉 | 0.8 | 4 |
| 牛奶 | 2.9 | 113 | 北豆腐 | 10.2 | 507 |
| 籼米 | 7.0 | 352 | 南豆腐 | 5.5 | 266 |
| 小麦粉 | 10.9 | 514 | 豆腐干 | 15.8 | 691 |
| 小米 | 9.3 | 510 | 瘦猪肉 | 17.3 | 805 |
| 白薯 | 1.0 | 51 | 瘦牛肉 | 19.0 | 700 |
| 土豆 | 2.1 | 70 | 鸡蛋 | 14.7 | 715 |
| 胡萝卜 | 0.9 | 17 | 水果 | 1.0 | —— |
2.皮肤护理 保持皮肤干燥,对皮肤皱褶处特别是腋下、腹股沟应保持清洁,有湿疹时应及时处理。
3.心理护理 为患儿家长讲解疾病知识,示范居家照护相关技能。评估家长的心理状态,为家长提供情感支持和心理疏导,缓解其负性情绪,建立照护患儿的信心,获得照护所需能力。
4.家庭支持 向家长提供诸如同伴支持、病友会、微信公众号等情感和信息支持,鼓励家长建立和维持正常的社会交往,以给予患儿足够的社会环境刺激,促进其心理社会发展。协助家长制订饮食治疗方案,提供遗传咨询;避免近亲结婚,所有新生儿出生数日后作常规筛查;有阳性家族史的新生儿生后应作详细检查;对患儿家族作苯丙氨酸耐量试验,检出杂合子。

